halfan (halofantrine hydrochloride) tablet
[GlaxoSmithKline]
WARNING
HALFAN HAS BEEN SHOWN TO PROLONG QTc INTERVAL AT THE RECOMMENDED THERAPEUTIC DOSE. THERE HAVE BEEN RARE REPORTS OF SERIOUS VENTRICULAR DYSRHYTHMIAS SOMETIMES ASSOCIATED WITH DEATH, WHICH MAY BE SUDDEN. HALFAN IS THEREFORE NOT RECOMMENDED FOR USE IN COMBINATION WITH DRUGS OR CLINICAL CONDITIONS KNOWN TO PROLONG QTc INTERVAL, OR IN PATIENTS WHO HAVE PREVIOUSLY RECEIVED MEFLOQUINE, OR IN PATIENTS WITH KNOWN OR SUSPECTED VENTRICULAR DYSRHYTHMIAS, A-V CONDUCTION DISORDERS OR UNEXPLAINED SYNCOPAL ATTACKS. HALFAN SHOULD BE PRESCRIBED ONLY BY PHYSICIANS WHO HAVE SPECIAL COMPETENCE IN THE DIAGNOSIS AND TREATMENT OF MALARIA, AND WHO ARE EXPERIENCED IN THE USE OF ANTIMALARIAL DRUGS. PHYSICIANS SHOULD THOROUGHLY FAMILIARIZE THEMSELVES WITH THE COMPLETE CONTENTS OF THIS LEAFLET BEFORE PRESCRIBING HALFAN.
DESCRIPTION
HALFAN (halofantrine hydrochloride) is an antimalarial drug available as tablets containing 250 mg of halofantrine hydrochloride (equivalent to 233 mg of the free base) for oral administration.
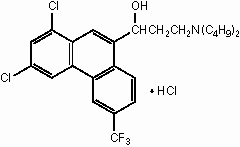
The chemical name of halofantrine hydrochloride is 1,3-dichloro-α-[2-(dibutylamino) ethyl]-6-(trifluoromethyl)-9-phenanthrene-methanol hydrochloride.
The drug, a white to off-white crystalline compound, is practically insoluble in water. Halofantrine hydrochloride has a calculated molecular weight of 536.89. The empirical formula is C26H30Cl2F3NO•HCl and the structural formula is
Inactive Ingredients
Inactive ingredients are magnesium stearate, microcrystalline cellulose, povidone, pregelatinized starch, sodium starch glycolate, and talc.
CLINICAL PHARMACOLOGY
The interindividual variability in the pharmacokinetic parameters of halofantrine is very wide and has led to great difficulty in precisely determining the pharmacokinetic characteristics of this product.
Following administration of halofantrine hydrochloride tablets in single oral doses of 250 mg to 1,000 mg to healthy volunteers, peak plasma levels were reached in 5 to 7 hours. High variability in the peak plasma levels was observed in all studies, suggesting erratic absorption from the gastrointestinal tract. An approximately 7-fold increase in peak plasma concentration and a 3-fold increase in area under the curve (AUC) of halofantrine were obtained when a single 250-mg tablet was administered with high-fat food to healthy subjects.
Healthy volunteers who were given 3 oral doses of 500 mg of halofantrine hydrochloride (500 mg every 6 hours), when fed 2 hours before the second and third doses, had similar 3- to 5-fold increases in absorption. A mean Cmax of 3,200 ng/mL (range 1,555 to 4,920 ng/mL) with a corresponding Tmax of 9 to 17 hours was attained following this multiple-dose regimen.
Halofantrine has a relatively long distribution phase with a half-life of 16 hours and a variable terminal elimination half-life of 6 to 10 days. The half-life of halofantrine varies considerably among individuals.
The primary metabolite of halofantrine is n-desbutyl halofantrine. Cmax valuesranging from 310 to 410 ng/mL were observed to occur between 32 and 56 hours following oral administration of multiple doses of 500 mg halofantrine q6h for 3 doses. The apparent terminal elimination half-life of the metabolite is 3 to 4 days.
Based on animal studies, hepatobiliary clearance with fecal elimination of halofantrine parent compound and metabolite predominates. The extent to which halofantrine is bound to plasma proteins and the extent to which halofantrine is taken up into red blood cells are unknown.
The pharmacokinetics of halofantrine in patients with compromised renal or hepatic function has not been investigated.
The course of the anemia developed by a few malaria patients treated with halofantrine whose red blood cells were deficient in glucose-6-phosphate dehydrogenase (G6PD) was not different from that in malaria patients with normal G6PD values.
Microbiology
Halofantrine is a blood schizonticidal antimalarial agent with no apparent action on the sporozoite, gametocyte, or hepatic stages of the infection. The exact mechanism of its action is unknown. The primary metabolite, n-desbutyl halofantrine, and the parent compound are equally active in vitro.
While in vitro studies indicate that there may be cross-resistance between halofantrine and mefloquine, the clinical data do not support this view. No significant correlation between halofantrine and mefloquine resistance was observed in clinical trials.
Clinical Trials
In controlled clinical trials involving 90 non-immune patients with malaria due to Plasmodium falciparum, treatment with HALFAN (500 mg every 6 hours for 3 doses on days 0 and 7) had a cure rate of 99%. Patients were followed for 28 days or more after initiation of treatment.
In trials involving 583 acute malaria patients, the majority of whom were semi-immune, treatment with HALFAN (500 mg every 6 hours for 3 doses) produced a cure rate of 90% against Plasmodium falciparum infection (n=512), and a cure rate of 99% against Plasmodium vivax (n=71).
INDICATIONS AND USAGE
HALFAN tablets are indicated for the treatment of adults who can tolerate oral medication and who have mild to moderate malaria (equal to or less than 100,000 parasites/mm3) caused by Plasmodium falciparum or Plasmodium vivax.
NOTE: Patients with acute P. vivax malaria treated with HALFAN are at risk of relapse because halofantrine does not eliminate the exoerythrocytic (hepatic phase) parasites. To avoid relapse after initial treatment of the acute P. vivax infection with HALFAN, patients should subsequently be treated with an 8-aminoquinoline to eradicate the exoerythrocytic parasites.
NOTE: THE EFFICACY OF HALFAN IN THE PROPHYLAXIS OF MALARIA HAS NOT BEEN ESTABLISHED.
CONTRAINDICATIONS
HALFANis contraindicated in patients with a known family history of congenital QTc prolongation. (See BOXED WARNING.) Use of this drug is contraindicated in patients with a known hypersensitivity to halofantrine.
WARNINGS
In life-threatening, severe, or overwhelming malarial infections, patients should be treated immediately with an appropriate parenteral antimalarial drug. The safety and efficacy of HALFAN in the treatment of patients with cerebral malaria or other forms of complicated malaria have not been established.
HALFAN has been shown to prolong QTc interval at the recommended therapeutic dose. There have been rare reports of serious ventricular dysrhythmias sometimes associated with death, which may be sudden. HALFAN is therefore not recommended in combination with drugs, or clinical conditions, known to prolong QTc interval, or in patients with known or suspected ventricular dysrhythmias, A-V conduction disorders, or unexplained syncopal attacks. Physicians should perform an ECG prior to dosing to ensure that the patient’s baseline QTc interval is within normal limits. Cardiac rhythm should be monitored during and for 8 to 12 hours following completion of therapy.
Caution should be used with concomitant intake of drugs which are known to significantly inhibit the hepatic cytochrome P450 enzyme, CYP3A4.
HALFAN should be taken on an empty stomach as increased absorption and, thus, increased toxicity may result from dosing in association with food. Do not exceed recommended doses, as higher than recommended doses of HALFAN have been shown to further prolong QTc interval.
Data on the use of HALFAN subsequent to administration of mefloquine suggest a significant, potentially fatal, prolongation of the QTc interval.1 Therefore, HALFAN should not be given simultaneously with, or subsequent to, mefloquine. (See PRECAUTIONS—Drug Interactions.)
Halofantrine has been shown to be embryotoxic in animal tests. Use in women of childbearing potential only with due caution regarding the potential effect on the fetus if the patient is pregnant. (See PRECAUTIONS—Pregnancy subsection.)
PRECAUTIONS
General
A phototoxic potential cannot be ruled out on the basis of the chemical moiety of halofantrine and the results of animal tests. (See ANIMAL TOXICOLOGY.) However, there is no evidence for this effect in humans.
Drug Interactions
Although no drug interaction studies have been conducted, HALFAN should not be administered with drugs known to prolong the QTc interval. In clinical use, an interaction with mefloquine has been reported to lead to further prolongation of the QTc interval.1 The prolongation may be significant and potentially fatal; therefore, HALFAN should not be given simultaneously with or subsequent to mefloquine. There have been no drug interactions reported when halofantrine is coadministered with chloroquine.
In vitro studies have shown that drugs which inhibit hepatic CYP3A4, e.g., ketoconazole, lead to an inhibition of halofantrine metabolism. Further, in dogs orally administered ketoconazole, the metabolism of halofantrine was decreased (SEE WARNINGS).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies of halofantrine hydrochloride in animals have not been performed to evaluate carcinogenic potential.
Genotoxicity of halofantrine hydrochloride was evaluated in 5 assay test systems including an Ames test, a gene mutation test in Chinese hamster ovary cells, a chromosomal aberration analysis in Chinese hamster ovary cells, a micronucleus test in mice, and a dominant lethal assay. No mutagenic potential was demonstrated in any of these test systems.
Halofantrine hydrochloride did not adversely affect male or female fertility in the rat at an oral dose of 30 mg/kg (1/6 of the maximum recommended human dose based on mg/m2).
Pregnancy
Teratogenic Effects
Pregnancy Category C: In pregnant rabbits, maternal-lethal doses (decremental dose schedule of 360 to 120 mg/kg, equivalent to 3.6 times to 1.2 times the maximum recommended human dose, respectively, based on mg/m2) were associated with abortion and an increased incidence of skeletal malformations, but oral doses up to 60 mg/kg (6/10 of the maximum recommended human dose based on mg/m2) did not produce maternal or fetal developmental toxicity.
Non-teratogenic Effects
In reproduction teratology studies in the rat, oral doses ≥30 g/kg (1/6 of the maximum recommended human dose based on mg/m2) produced postimplantation embryonic death and reduced fetal weight and viability. Halofantrine hydrochloride at doses of 15 mg/kg/day (1/10 of the maximum recommended human dose based on mg/m2) had no embryotoxicity or teratogenicity. These effects occurred at and below doses that produced overt maternal toxicity in the rats.
Halofantrine has been shown to be embryocidal in rats. There are no adequate and well-controlled studies in pregnant women. HALFAN should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
In lactation studies performed in rats, dose-related decreases in offspring body weight were observed at a dose of 25 mg/kg/day and above (1/8 of the maximum recommended human dose based on mg/m2). Control pups breast-fed by high-dosed mothers had significant decreases in body weight and survival at doses of 50 and 100 mg/kg/day (1/4 to 1/2 of the maximum recommended human dose based on mg/m2).
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from halofantrine hydrochloride, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness of halofantrine hydrochloride tablets in the pediatric population have not been established.
Geriatric Use
There are no studies on the use of halofantrine hydrochloride in elderly individuals.
ADVERSE REACTIONS
Normal Subjects
The following adverse events were reported in normal subjects given HALFAN 1,000 mg to 1,500 mg in a single dosing course.
Gastrointestinal
Abdominal pain (10%), anorexia (5%), diarrhea (5%), nausea (10%), vomiting (10%).
Central Nervous System
Dizziness (5%), headache (5%).
Clinical Trials
In clinical trials involving 933 patients treated with three 500 mg doses (500 mg every 6 hours), the following clinical adverse events were reported.
There were no deaths or permanent disabilities thought related to drug toxicity. Five patients discontinued medication due to adverse events. Three patients vomited medicine repeatedly.
Though temporally related to drug administration, the relationship of the following serious adverse events to malaria or underlying illness as opposed to drug toxicity could not be established. Two patients had decreased consciousness; other serious adverse events reported during clinical trials included convulsions (3 cases), stomatitis (3 cases), moderately severe diarrhea (2 cases), pulmonary edema (1 case), tetany (1 case), hypertensive crisis (1 case), cerebrovascular accident (1 case).
The most frequently reported adverse events thought possibly— or probably—related to halofantrine were: Abdominal pain (8.5%), diarrhea (6.0%), dizziness (4.5%), vomiting (4.3%), nausea (3.4%), cough (3.0%), headache (3.0%), pruritus (2.6%), rigors (1.7%), and myalgias (1.3%). These events are also characteristic of malaria.
Pruritus was reported in a higher proportion of highly pigmented patients than in other patients.
Adverse events thought possibly—or probably—related to halofantrine affecting <1% of patients studied in the clinical trials included:
Body as a Whole
Fatigue, malaise.
Cardiovascular
Chest pain, palpitations, postural hypotension.
Dermatologic
Rash.
Gastrointestinal
Abdominal distention, anorexia, constipation, dyspepsia.
Mucous Membrane
Stomatitis.
Musculoskeletal
Arthralgia, back pain.
Central Nervous System
Asthenia, confusion, convulsions, depression, paresthesia, sleep disorder.
Renal
Urinary frequency.
Special Senses
Abnormal vision, tinnitus.
Laboratory
The most frequently noted laboratory abnormalities that occurred following drug administration in the clinical trials were decreased hematocrit, elevated hepatic transaminases, decreased and increased white blood cell counts, and decreased platelet counts. These alterations returned to normal limits within 2 to 3 weeks post-infection. The causal relationship of these changes to HALFAN is unclear, as these laboratory abnormalities can also occur with acute malaria.
Postmarketing Experience
Halofantrine was marketed in Europe starting in 1988. The following additional adverse experiences have been reported in postmarketing surveillance outside the United States: Facial edema and urticaria (allergic/anaphylactic reactions) in rare cases.
Hemolysis/hemolytic anemia (including immune hemolytic anemia) which may compromise renal function have been reported in patients with malaria who have been treated with halofantrine. Hemolytic reactions may also be observed in patients with malaria in the absence of halofantrine.
Prolongation of QT interval has been reported. There have been rare reports of serious ventricular dysrhythmias sometimes associated with death. These cases have occurred particularly under certain conditions which include uses of doses higher than recommended, recent or concomitant treatment with mefloquine, or presence of pre-existing prolongation of QT interval.1
OVERDOSAGE
In case of overdosage, vomiting should be induced, in conjunction with appropriate supportive measures, which should include ECG monitoring. The possibility of neurologic toxicity, especially decreased consciousness and seizures, should be evaluated. Dehydration secondary to gastrointestinal toxicity with diarrhea and vomiting may require treatment with intravenous fluid therapy.
Gastrointestinal distress with abdominal pain, vomiting, cramping, and diarrhea occurs at doses higher than the recommended therapeutic regimen. Palpitations have also been reported at these higher doses.
DOSAGE AND ADMINISTRATION
(See INDICATIONS AND USAGE.)
HALFAN should be given on an empty stomach at least 1 hour before or 2 hours after food. (See WARNINGS.) The recommended dosage regimen to treat adults able to tolerate oral medications, who have mild to moderate malaria caused by P. falciparum or P. vivax is:
Non-immune Patients
Patients with no previous exposure or minimal exposure to malaria should be considered “non-immune.” These patients should receive 500 mg (2 x 250 mg tablets) of halofantrine hydrochloride every 6 hours for 3 doses (total first course dosage 1,500 mg). This course of therapy should be repeated 7 days after the first course.
Semi-immune Patients
Patients with a history of life-long residence in endemic areas and a clear history of recent previous malaria caused by the same Plasmodium species may be considered semi-immune. In these patients, omitting the second course of therapy may be considered. Clinical trials in semi-immune patients have utilized this one-course regimen with satisfactory efficacy and safety.
Hepatic or Renally Impaired Patients
There is no information on dosing alterations needed because of hepatic or renal impairment.
HOW SUPPLIED
HALFAN is available as a white to off-white, capsule-shaped tablet containing 250 mg of halofantrine hydrochloride in bottles of 60 tablets. The tablets are imprinted HALFAN on 1 side.
Store at controlled room temperature between 20° to 25°C (68° to 77°F) and protect from light.
250 mg 60’s: NDC 0007-4195-18
ANIMAL TOXICOLOGY
In a phototoxicity study, halofantrine hydrochloride was phototoxic to mice at 80 mg/kg (6/10 of the maximum recommended human dose based on mg/m2). At 40 mg/kg, the lowest dose tested, there was a slight erythematous response.
In a whole-body radioautographic study in the rat, it was demonstrated that high drug levels of halofantrine are retained in the retina and in the Harderian gland for an entire 4-week observation period. Moreover, the estimated half-lives for the radiolabeled equivalents in the retina and Harderian gland were from 91 to 778 hours for the 4-week observation period. The drug passes the blood-brain barrier and is retained for an undetermined time in the central nervous system.
Elevated cholesterol values have been reported in the rat when halofantrine hydrochloride is administered for 4 weeks at oral doses of 30 mg/kg (2/10 of the maximum recommended human dose based on mg/m2) and higher.
Increases in serum cholesterol have also been reported in dogs administered halofantrine hydrochloride for 28 days at oral doses of 25 mg/kg (1/2 of the recommended human dose based on mg/m2) and higher.
REFERENCES
- Nosten F, ter Kuile FO, Luxemburger C, et al. Cardiac effects of antimalarial treatment with halofantrine. Lancet. 1993;341:1054-56.
DATE OF ISSUANCE JUNE 2003
©2003, GlaxoSmithKline
Manufactured by King Pharmaceuticals, Inc.
Bristol, TN 37620
for
GlaxoSmithKline
Research Triangle Park NC 27709
HL:L5
| HALFAN (halofantrine hydrochloride) | ||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
Revised: 09/2006