ketek (telithromycin) tablet, film coated
[Sanofi-Aventis U.S. LLC]
To reduce the development of drug-resistant bacteria and maintain the effectiveness of KETEK and other antibacterial drugs, KETEK should be used only to treat infections that are proven or strongly suspected to be caused by bacteria.
DESCRIPTION
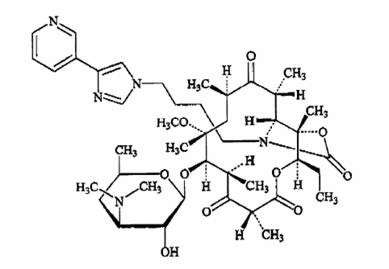
KETEK® tablets contain telithromycin, a semisynthetic antibacterial in the ketolide class for oral administration. Chemically, telithromycin is designated as Erythromycin, 3-de[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-11,12-dideoxy-6-O-methyl-3-oxo-12,11-[oxycarbonyl[[4-[4-(3-pyridinyl)-1H-imidazol-1-yl]butyl]imino]]-.
Telithromycin, a ketolide, differs chemically from the macrolide group of antibacterials by the lack of α-L-cladinose at position 3 of the erythronolide A ring, resulting in a 3-keto function. It is further characterized by a C11-12 carbamate substituted by an imidazolyl and pyridyl ring through a butyl chain. Its empirical formula is C43H65N5O10 and its molecular weight is 812.03. Telithromycin is a white to off-white crystalline powder. The following represents the chemical structure of telithromycin.
KETEK tablets are available as light-orange, oval, film-coated tablets, each containing 400 mg or 300 mg of telithromycin, and the following inactive ingredients: croscarmellose sodium, hypromellose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, povidone, red ferric oxide, talc, titanium dioxide, and yellow ferric oxide.
CLINICAL PHARMACOLOGY
Pharmacokinetics
Absorption
Following oral administration, telithromycin reached maximal concentration at about 1 hour (0.5 – 4 hours).
It has an absolute bioavailability of 57% in both young and elderly subjects.
The rate and extent of absorption are unaffected by food intake, thus KETEK tablets can be given without regard to food.
In healthy adult subjects, peak plasma telithromycin concentrations of approximately 2 µg/mL are attained at a median of 1 hour after an 800-mg oral dose.
Steady-state plasma concentrations are reached within 2 to 3 days of once daily dosing with telithromycin 800 mg.
Following oral dosing, the mean terminal elimination half-life of telithromycin is 10 hours.
The pharmacokinetics of telithromycin after administration of single and multiple (7 days) once daily 800-mg doses to healthy adult subjects are shown in Table 1.
| Mean (SD) | ||
|---|---|---|
| Parameter | Single dose (n=18) | Multiple dose (n=18) |
| SD=Standard deviation | ||
| Cmax=Maximum plasma concentration | ||
| Tmax=Time to Cmax | ||
| AUC=Area under concentration vs. time curve | ||
| t1/2=Terminal plasma half-life | ||
| C24h=Plasma concentration at 24 hours post-dose | ||
|
||
| Cmax (µg/mL) | 1.9 (0.80) | 2.27 (0.71) |
| Tmax (h)* | 1.0 (0.5–4.0) | 1.0 (0.5–3.0) |
| AUC(0–24) (µg∙h/mL) | 8.25 (2.6) | 12.5 (5.4) |
| Terminal t1/2 (h) | 7.16 (1.3) | 9.81 (1.9) |
| C24h (µg/mL) | 0.03 (0.013) | 0.07 (0.051) |
In a patient population, mean peak and trough plasma concentrations were 2.9 µg/mL (±1.55), (n=219) and 0.2 µg/mL (±0.22), (n=204), respectively, after 3 to 5 days of KETEK 800 mg once daily.
Distribution
Total in vitro protein binding is approximately 60% to 70% and is primarily due to human serum albumin.
Protein binding is not modified in elderly subjects and in patients with hepatic impairment.
The volume of distribution of telithromycin after intravenous infusion is 2.9 L/kg.
Telithromycin concentrations in bronchial mucosa, epithelial lining fluid, and alveolar macrophages after 800 mg once daily dosing for 5 days in patients are displayed in Table 2.
| Hours post-dose |
Mean concentration (µg/mL) | Tissue/Plasma Ratio | ||
|---|---|---|---|---|
| Tissue or fluid |
Plasma | |||
|
||||
| Bronchial mucosa | 2 | 3.88* | 1.86 | 2.11 |
| 12 | 1.41* | 0.23 | 6.33 | |
| 24 | 0.78* | 0.08 | 12.11 | |
| Epithelial lining fluid | 2 | 14.89 | 1.86 | 8.57 |
| 12 | 3.27 | 0.23 | 13.8 | |
| 24 | 0.84 | 0.08 | 14.41 | |
| Alveolar macrophages | 2 | 65 | 1.07 | 55 |
| 8 | 100 | 0.605 | 180 | |
| 24 | 41 | 0.073 | 540 | |
Telithromycin concentration in white blood cells exceeds the concentration in plasma and is eliminated more slowly from white blood cells than from plasma. Mean white blood cell concentrations of telithromycin peaked at 72.1µg/mL at 6 hours, and remained at 14.1 µg/mL 24 hours after 5 days of repeated dosing of 600 mg once daily. After 10 days, repeated dosing of 600 mg once daily, white blood cell concentrations remained at 8.9 µg/mL 48 hours after the last dose.
Metabolism
In total, metabolism accounts for approximately 70% of the dose. In plasma, the main circulating compound after administration of an 800-mg radiolabeled dose was parent compound, representing 56.7% of the total radioactivity. The main metabolite represented 12.6% of the AUC of telithromycin. Three other plasma metabolites were quantified, each representing 3% or less of the AUC of telithromycin.
It is estimated that approximately 50% of its metabolism is mediated by CYP 450 3A4 and the remaining 50% is CYP 450-independent.
Elimination
The systemically available telithromycin is eliminated by multiple pathways as follows: 7% of the dose is excreted unchanged in feces by biliary and/or intestinal secretion; 13% of the dose is excreted unchanged in urine by renal excretion; and 37% of the dose is metabolized by the liver.
Special populations
Gender
There was no significant difference between males and females in mean AUC, Cmax, and elimination half-life in two studies; one in 18 healthy young volunteers (18 to 40 years of age) and the other in 14 healthy elderly volunteers (65 to 92 years of age), given single and multiple once daily doses of 800 mg of KETEK.
Hepatic insufficiency
In a single-dose study (800 mg) in 12 patients and a multiple-dose study (800 mg) in 13 patients with mild to severe hepatic insufficiency (Child Pugh Class A, B and C), the Cmax, AUC and t1/2 of telithromycin were similar to those obtained in age- and sex-matched healthy subjects. In both studies, an increase in renal elimination was observed in hepatically impaired patients indicating that this pathway may compensate for some of the decrease in metabolic clearance. No dosage adjustment is recommended due to hepatic impairment. (See PRECAUTIONS, General and DOSAGE AND ADMINISTRATION.)
Renal insufficiency
In a multiple-dose study, 36 subjects with varying degrees of renal impairment received 400 mg, 600 mg, or 800 mg KETEK once daily for 5 days. There was a 1.4-fold increase in Cmax,ss, and a 1.9-fold increase in AUC (0–24)ss at 800 mg multiple doses in the severely renally impaired group (CLCR< 30 mL/min) compared to healthy volunteers. Renal excretion may serve as a compensatory elimination pathway for telithromycin in situations where metabolic clearance is impaired. Patients with severe renal impairment are prone to conditions that may impair their metabolic clearance. Therefore, in the presence of severe renal impairment (CLCR< 30 mL/min), a reduced dosage of KETEK is recommended. (See DOSAGE AND ADMINISTRATION.)
In a single-dose study in patients with end-stage renal failure on hemodialysis (n=10), the mean Cmax and AUC values were similar to normal healthy subjects when KETEK was administered 2 hours post-dialysis. However, the effect of dialysis on removing telithromycin from the body has not been studied.
Multiple insufficiency
The effects of co-administration of ketoconazole in 12 subjects (age ≥ 60 years), with impaired renal function were studied (CLCR= 24 to 80 mL/min). In this study, when severe renal insufficiency(CLCR< 30 mL/min, n=2) and concomitant impairment of CYP 3A4 metabolism pathway were present, telithromycin exposure (AUC (0–24)) was increased by approximately 4- to 5-fold compared with the exposure in healthy subjects with normal renal function receiving telithromycin alone. In the presence of severe renal impairment (CLCR< 30 mL/min), with coexisting hepatic impairment, a reduced dosage of KETEK is recommended. (See PRECAUTIONS, General and DOSAGE AND ADMINISTRATION.)
Geriatric
Pharmacokinetic data show that there is an increase of 1.4-fold in exposure (AUC) in 20 patients ≥ 65 years of age with community acquired pneumonia in a Phase III study, and a 2.0-fold increase in exposure (AUC) in 14 subjects ≥ 65 years of age as compared with subjects less than 65 years of age in a Phase I study. No dosage adjustment is required based on age alone.
Drug-drug interactions
Studies were performed to evaluate the effect of CYP 3A4 inhibitors on telithromycin and the effect of telithromycin on drugs that are substrates of CYP 3A4 and CYP 2D6. In addition, drug interaction studies were conducted with several other concomitantly prescribed drugs.
CYP 3A4 inhibitors
Itraconazole
A multiple-dose interaction study with itraconazole showed that Cmax of telithromycin was increased by 22% and AUC by 54%.
Ketoconazole
A multiple-dose interaction study with ketoconazole showed that Cmax of telithromycin was increased by 51% and AUC by 95%.
Grapefruit juice
When telithromycin was given with 240 mL of grapefruit juice after an overnight fast to healthy subjects, the pharmacokinetics of telithromycin were not affected.
CYP 3A4 substrates
Cisapride
Steady-state peak plasma concentrations of cisapride (an agent with the potential to increase QT interval) were increased by 95% when co-administered with repeated doses of telithromycin, resulting in significant increases in QTc. (See CONTRAINDICATIONS.)
Simvastatin
When simvastatin was co-administered with telithromycin, there was a 5.3-fold increase in simvastatin Cmax, an 8.9-fold increase in simvastatin AUC, a 15-fold increase in the simvastatin active metabolite Cmax, and a 12-fold increase in the simvastatin active metabolite AUC. (See PRECAUTIONS.)
In another study, when simvastatin and telithromycin were administered 12 hours apart, there was a 3.4-fold increase in simvastatin Cmax, a 4.0-fold increase in simvastatin AUC, a 3.2-fold increase in the active metabolite Cmax, and a 4.3-fold increase in the active metabolite AUC. (See PRECAUTIONS.)
Midazolam
Concomitant administration of telithromycin with intravenous or oral midazolam resulted in 2- and 6-fold increases, respectively, in the AUC of midazolam due to inhibition of CYP 3A4-dependent metabolism of midazolam. (See PRECAUTIONS.)
CYP 2D6 substrates
Paroxetine
There was no pharmacokinetic effect on paroxetine when telithromycin was co-administered.
Metoprolol
When metoprolol was co-administered with telithromycin, there was an increase of approximately 38% on the Cmax and AUC of metoprolol, however, there was no effect on the elimination half-life of metoprolol. Telithromycin exposure is not modified with concomitant single-dose administration of metoprolol. (See PRECAUTIONS, Drug interactions.)
Other drug interactions
Digoxin
The plasma peak and trough levels of digoxin were increased by 73% and 21%, respectively, in healthy volunteers when co-administered with telithromycin. However, trough plasma concentrations of digoxin (when equilibrium between plasma and tissue concentrations has been achieved) ranged from 0.74 to 2.17 ng/mL. There were no significant changes in ECG parameters and no signs of digoxin toxicity. (See PRECAUTIONS.)
Theophylline
When theophylline was co-administered with repeated doses of telithromycin, there was an increase of approximately 16% and 17% on the steady-state Cmax and AUC of theophylline. Co-administration of theophylline may worsen gastrointestinal side effects such as nausea and vomiting, especially in female patients. It is recommended that telithromycin should be taken with theophylline 1 hour apart to decrease the likelihood of gastrointestinal side effects.
Sotalol
Telithromycin has been shown to decrease the Cmax and AUC of sotalol by 34% and 20%, respectively, due to decreased absorption.
Warfarin
When co-administered with telithromycin in healthy subjects, there were no pharmacodynamic or pharmacokinetic effects on racemic warfarin.
Oral contraceptives
When oral contraceptives containing ethinyl estradiol and levonorgestrel were co-administered with telithromycin, the steady-state AUC of ethinyl estradiol did not change and the steady-state AUC of levonorgestrel was increased by 50%. The pharmacokinetic/pharmacodynamic study showed that telithromycin did not interfere with the antiovulatory effect of oral contraceptives containing ethinyl estradiol and levonorgestrel.
Ranitidine, antacid
There was no clinically relevant pharmacokinetic interaction of ranitidine or antacids containing aluminum and magnesium hydroxide on telithromycin.
Rifampin
During concomitant administration of rifampin and KETEK in repeated doses, Cmax and AUC of telithromycin were decreased by 79%, and 86%, respectively. (See PRECAUTIONS, Drug Interactions.)
Microbiology
Telithromycin belongs to the ketolide class of antibacterials and is structurally related to the macrolide family of antibiotics. Telithromycin concentrates in phagocytes where it exhibits activity against intracellular respiratory pathogens. In vitro, telithromycin has been shown to demonstrate concentration-dependent bactericidal activity against isolates of Streptococcus pneumoniae (including multi-drug resistant isolates [MDRSP*]).
*MDRSP=Multi-drug resistant Streptococcus pneumoniae includes isolates known as PRSP (penicillin-resistant Streptococcus pneumoniae), and are isolates resistant to two or more of the following antimicrobials: penicillin, 2nd generation cephalosporins (e.g., cefuroxime), macrolides, tetracyclines, and trimethoprim/sulfamethoxazole.
Mechanism of action
Telithromycin blocks protein synthesis by binding to domains II and V of 23S rRNA of the 50S ribosomal subunit. By binding at domain II, telithromycin retains activity against gram-positive cocci (e.g., Streptococcus pneumoniae) in the presence of resistance mediated by methylases (erm genes) that alter the domain V binding site of telithromycin. Telithromycin may also inhibit the assembly of nascent ribosomal units.
Mechanism of resistance
Staphylococcus aureus and Streptococcus pyogenes with the constitutive macrolide-lincosamide-streptogramin B (cMLSB) phenotype are resistant to telithromycin.
Mutants of Streptococcus pneumoniae derived in the laboratory by serial passage in subinhibitory concentrations of telithromycin have demonstrated resistance based on L22 riboprotein mutations (telithromycin MICs are elevated but still within the susceptible range), one of two reported mutations affecting the L4 riboprotein, and production of K-peptide. The clinical significance of these laboratory mutants is not known.
Cross resistance
Telithromycin does not induce resistance through methylase gene expression in erythromycin-inducibly resistant bacteria, a function of its 3-keto moiety. Telithromycin has not been shown to induce resistance to itself.
List of Microorganisms
Telithromycin has been shown to be active against most strains of the following microorganisms, both in vitro and in clinical settings as described in the INDICATIONS AND USAGE section.
Aerobic gram-positive microorganisms
- Staphylococcus aureus (methicillin and erythromycin susceptible isolates only)
- Streptococcus pneumoniae (including multi-drug resistant isolates [MDRSP*])
*MDRSP=Multi-drug resistant Streptococcus pneumoniae includes isolates known as PRSP (penicillin-resistant S. pneumoniae), and are isolates resistant to two or more of the following antimicrobials: penicillin, 2nd generation cephalosporins (e.g., cefuroxime), macrolides, tetracyclines, and trimethoprim/sulfamethoxazole.
Aerobic gram-negative microorganisms
- Haemophilus influenzae
- Moraxella catarrhalis
Other microorganisms
- Chlamydophila (Chlamydia) pneumoniae
- Mycoplasma pneumoniae
The following in vitro data are available, but their clinical significance is unknown.
At least 90% of the following microorganisms exhibit in vitro minimum inhibitory concentrations (MICs) less than or equal to the susceptible breakpoint for telithromycin. However, the safety and efficacy of telithromycin in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled clinical trials.
Aerobic gram-positive microorganisms
- Streptococcus pyogenes (erythromycin susceptible isolates only)
- Streptococci (Lancefield groups C and G)
- Viridans group streptococci
Anaerobic bacteria
- Prevotella bivia
- Prevotella intermedia
- Peptostreptococcus spp.
Other microorganisms
- Legionella pneumophila
Susceptibility Test Methods
When available, the clinical microbiology laboratory should provide cumulative results of in vitro susceptibility test results for antimicrobial drugs used in local hospitals and practice areas to the physician as periodic reports that describe the susceptibility profile of nosocomial and community-acquired pathogens. These reports should aid the physician in selecting the most effective antimicrobial.
Dilution techniques
Quantitative methods are used to determine antimicrobial minimum inhibitory concentrations (MICs). These MICs provide estimates of the susceptibility of bacteria to antibacterial compounds. The MICs should be determined using a standardized procedure. Standardized procedures are based on dilution methods (broth or agar dilution)1,3 or equivalent with standardized inoculum and concentrations of telithromycin powder. The MIC values should be interpreted according to criteria provided in Table 3.
Diffusion techniques
Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antibiotics. One such standardized procedure2,3 requires the use of standardized inoculum concentrations. This procedure uses paper disks impregnated with 15 µg telithromycin to test the susceptibility of microorganisms to telithromycin. Disc diffusion zone sizes should be interpreted according to criteria in Table 3.
| Minimal Inhibitory Concentrations (µg/mL) |
Disk Diffusion (zone diameters in mm) |
|||||
|---|---|---|---|---|---|---|
| Pathogen | S | I | R* | S | I | R* |
|
||||||
| Staphylococcus aureus |
≤ 0.25 | ≥ 22 | ||||
| Streptococcus pneumoniae |
≤ 1 | 2 | ≥ 4 | ≥ 19 | 16–18 | ≤ 15 |
| Haemophilus influenzae |
≤ 4 | 8 | ≥ 16 | ≥ 15 | 12–14 | ≤ 11 |
A report of "Susceptible" indicates that the antimicrobial is likely to inhibit growth of the pathogen if the antibacterial compound in the blood reaches the concentrations usually achievable. A report of "Intermediate" indicates that the result should be considered equivocal, and, if the microorganism is not fully susceptible to alternative, clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body sites where the drug is physiologically concentrated or in situations where high dosage of drug can be used. This category also provides a buffer zone that prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of "Resistant" indicates that the antimicrobial is not likely to inhibit growth of the pathogen if the antimicrobial compound in the blood reaches the concentrations usually achievable; other therapy should be selected.
Quality control
Standardized susceptibility test procedures require the use of quality control microorganisms to determine the performance of the test procedures1,2,3. Standard telithromycin powder should provide the MIC ranges for the quality control organisms in Table 4. For the disk diffusion technique, the 15-µg telithromycin disk should provide the zone diameter ranges for the quality control organisms in Table 4.
| QC Strain | Minimum Inhibitory Concentrations (µg/mL) |
Disk Diffusion (Zone diameter in mm) |
|---|---|---|
| ATCC = American Type Culture Collection | ||
| Staphylococcus aureus ATCC® 29213 |
0.06–0.25 | Not Applicable |
| Staphylococcus aureus ATCC 25923 |
Not Applicable | 24–30 |
| Streptococcus pneumoniae ATCC 49619 |
0.004–0.03 | 27–33 |
| Haemophilus influenzae ATCC 49247 |
1.0–4.0 | 17–23 |
INDICATIONS AND USAGE
KETEK tablets are indicated for the treatment of infections caused by susceptible strains of the designated microorganisms in the conditions listed below for patients 18 years old and above.
Acute bacterial exacerbation of chronic bronchitis due to Streptococcus pneumoniae, Haemophilus influenzae, or Moraxella catarrhalis.
Acute bacterial sinusitis due to Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis, or Staphylococcus aureus.
Community-acquired pneumonia (of mild to moderate severity) due to Streptococcus pneumoniae, (including multi-drug resistant isolates [MDRSP*]), Haemophilus influenzae, Moraxella catarrhalis, Chlamydophila pneumoniae, or Mycoplasma pneumoniae.
*MDRSP, Multi-drug resistant Streptococcus pneumoniae includes isolates known as PRSP (penicillin-resistant Streptococcus pneumoniae), and are isolates resistant to two or more of the following antibiotics: penicillin, 2nd generation cephalosporins, e.g., cefuroxime, macrolides, tetracyclines and trimethoprim/sulfamethoxazole.
To reduce the development of drug-resistant bacteria and maintain the effectiveness of KETEK and other antibacterial drugs, KETEK should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
CONTRAINDICATIONS
KETEK is contraindicated in patients with a history of hypersensitivity to telithromycin and/or any components of KETEK tablets, or any macrolide antibiotic.
KETEK is contraindicated in patients with previous history of hepatitis and/or jaundice associated with the use of KETEK tablets, or any macrolide antibiotic.
Concomitant administration of KETEK with cisapride or pimozide is contraindicated. (See CLINICAL PHARMACOLOGY, Drug-drug Interactions and PRECAUTIONS.)
WARNINGS
Hepatotoxicity
Acute hepatic failure and severe liver injury, in some cases fatal, have been reported in patients treated with KETEK. These hepatic reactions included fulminant hepatitis and hepatic necrosis leading to liver transplant, and were observed during or immediately after treatment. In some of these cases, liver injury progressed rapidly and occurred after administration of a few doses of KETEK. (See ADVERSE REACTIONS.)
Physicians and patients should monitor for the appearance of signs or symptoms of hepatitis, such as fatigue, malaise, anorexia, nausea, jaundice, bilirubinuria, acholic stools, liver tenderness or hepatomegaly. Patients with signs or symptoms of hepatitis mustbe advised to discontinue KETEK and immediately seek medical evaluation, which should include liver function tests. (See ADVERSE REACTIONS, PRECAUTIONS, Information to Patients.) If clinical hepatitis or transaminase elevations combined with other systemic symptoms occur, KETEK should be permanently discontinued.
Ketek must not be re-administered to patients with a previous history of hepatitis and/or jaundice associated with the use of KETEK tablets, or any macrolide antibiotic. (See CONTRAINDICATIONS.)
Exacerbations of myasthenia gravis
Telithromycin should not be used in patients with myasthenia gravis unless no other therapeutic alternatives are available. Exacerbations of myasthenia gravis have been reported in patients with myasthenia gravis treated with telithromycin. This has sometimes occurred within a few hours after intake of the first dose of telithromycin. Reports have included death and life-threatening acute respiratory failure with a rapid onset in patients with myasthenia gravis treated for respiratory tract infections with telithromycin. If other therapeutic alternatives are not available, patients with myasthenia gravis taking telithromycin must be closely monitored. Patients must be advised that if they experience exacerbation of their symptoms, they should discontinue treatment of KETEK and immediately seek medical attention. Supportive measures should be instituted as medically necessary.
QTc interval prolongation
Telithromycin has the potential to prolong the QTc interval of the electrocardiogram in some patients. QTc prolongation may lead to an increased risk for ventricular arrhythmias, including torsades de pointes. Thus, telithromycin should be avoided in patients with congenital prolongation of the QTc interval, and in patients with ongoing proarrhythmic conditions such as uncorrected hypokalemia or hypomagnesemia, clinically significant bradycardia, and in patients receiving Class IA (e.g., quinidine and procainamide) or Class III (e.g., dofetilide) antiarrhythmic agents.
No cardiovascular morbidity or mortality attributable to QTc prolongation occurred with telithromycin treatment in 4780 patients in clinical efficacy trials, including 204 patients having a prolonged QTc at baseline.
Pseudomembranous colitis
Pseudomembranous colitis has been reported with nearly all antibacterial agents, including telithromycin, and may range in severity from mild to life-threatening. Therefore, it is important to consider this diagnosis in patients who present with diarrhea subsequent to the administration of any antibacterial agents.
Treatment with antibacterial agents alters the flora of the colon and may permit overgrowth of clostridia. Studies indicate that toxin-producing strains of Clostridium difficile are the primary cause of "antibiotic-associated colitis".
After the diagnosis of pseudomembranous colitis has been established, therapeutic measures should be initiated. Mild cases of pseudomembranous colitis usually respond to drug discontinuation alone. In moderate to severe cases, consideration should be given to management with fluids and electrolytes, protein supplementation, and treatment with an antibacterial drug clinically effective against C. difficile colitis. (See ADVERSE REACTIONS.)
PRECAUTIONS
General
Prescribing KETEK in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
KETEK may cause visual disturbances particularly in slowing the ability to accommodate and the ability to release accommodation. Visual disturbances included blurred vision, difficulty focusing, and diplopia. Most events were mild to moderate; however, severe cases have been reported.
There have been post-marketing adverse event reports of syncope usually associated with vagal syndrome.
Patients should be cautioned about the potential effects of these visual disturbances and syncope on driving a vehicle, operating machinery or engaging in other potentially hazardous activities. (See ADVERSE REACTIONS, CLINICAL STUDIES.)
Hepatic dysfunction, including increased liver enzymes and hepatitis, with or without jaundice, has been reported with the use of KETEK. These events were generally reversible, though acute hepatic failure and severe liver injury, in some cases fatal, have been reported.(See WARNINGS, ADVERSE REACTIONS, Liver and biliary system.)
Telithromycin is principally excreted via the liver and kidney. Telithromycin may be administered without dosage adjustment in the presence of hepatic impairment. In the presence of severe renal impairment (CLCR< 30 mL/min), a reduced dosage of KETEK is recommended. (See DOSAGE AND ADMINISTRATION.)
Information for patients
The following information and instructions should be communicated to the patient.
KETEK may cause problems with vision particularly when looking quickly between objects close by and objects far away. These events include blurred vision, difficulty focusing, and objects looking doubled. Most events were mild to moderate; however, severe cases have been reported. Problems with vision were reported as having occurred after any dose during treatment, but most occurred following the first or second dose. These problems lasted several hours and in some patients came back with the next dose. (See PRECAUTIONS, General and ADVERSE REACTIONS.)
If visual difficulties occur:
- patients should avoid driving a motor vehicle, operating heavy machinery, or engaging in otherwise hazardous activities.
- avoiding quick changes in viewing between objects in the distance and objects nearby may help to decrease the effects of these visual difficulties.
- patients should contact their physician if these visual difficulties interfere with their daily activities.
Patients should be aware of the possibility of experiencing syncope (fainting), and its impact on the ability to drive, especially if they are experiencing vagal symptoms (severe nausea, vomiting, and/or lightheadedness).
If patients experience these symptoms, they should avoid driving a motor vehicle, operating heavy machinery, or engaging in otherwise hazardous activities.
Patients should also be advised:
- of the possibility of severe liver injury, associated with KETEK, which in rare cases may be severe. Patients developing signs or symptoms of liver injury should be instructed to discontinue KETEK and seek medical attention immediately. Symptoms of liver injury may include nausea, fatigue, anorexia, jaundice, dark urine, light-colored stools, pruritus, or tender abdomen. KETEK must not be taken by patients with a previous history of hepatitis/jaundice associated with the use of KETEK. (See CONTRAINDICATIONS and WARNINGS.)
- Patients with myasthenia gravis should not take KETEK , unless there are no other therapeutic alternatives. Exacerbations of myasthenia gravis have been reported in patients treated with KETEK. This has sometimes occurred within a few hours after taking the first dose. Reports have included death and life-threatening respiratory failure that occurred rapidly in patients with myasthenia gravis. (See WARNINGS).
- that antibacterial drugs including KETEK should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When KETEK is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by KETEK or other antibacterial drugs in the future.
- that KETEK has the potential to produce changes in the electrocardiogram (QTc interval prolongation) and that they should report any fainting occurring during drug treatment.
- that KETEK should be avoided in patients receiving Class 1A (e.g., quinidine, procainamide) or Class III (e.g., dofetilide) antiarrhythmic agents.
- to inform their physician of any personal or family history of QTc prolongation or proarrhythmic conditions such as uncorrected hypokalemia, or clinically significant bradycardia.
- that simvastatin, lovastatin, or atorvastatin should be avoided in patients receiving KETEK. If KETEK is prescribed, therapy with simvastatin, lovastatin, or atorvastatin should be stopped during the course of treatment.
- that KETEK tablets can be taken with or without food.
- to inform their physician of any other medications taken concurrently with KETEK, including over-the-counter medications and dietary supplements.
Drug interactions
Telithromycin is a strong inhibitor of the cytochrome P450 3A4 system. Co-administration of KETEK tablets and a drug primarily metabolized by the cytochrome P450 3A4 enzyme system may result in increased plasma concentration of the drug co-administered with telithromycin that could increase or prolong both the therapeutic and adverse effects. Therefore, appropriate dosage adjustments may be necessary for the drug co-administered with telithromycin.
The use of KETEK is contraindicated with cisapride. (See CONTRAINDICATIONS and CLINICAL PHARMACOLOGY, Drug-drug interactions.)
The use of KETEK is contraindicated with pimozide. Although there are no studies looking at the interaction between KETEK and pimozide, there is a potential risk of increased pimozide plasma levels by inhibition of CYP 3A4 pathways by KETEK as with macrolides. (See CONTRAINDICATIONS.)
In a pharmacokinetic study, simvastatin levels were increased due to CYP 3A4 inhibition by telithromycin. (See CLINICAL PHARMACOLOGY, Other drug interactions.) Similarly, an interaction may occur with lovastatin or atorvastatin, but not with pravastatin or fluvastatin. High levels of HMG-CoA reductase inhibitors increase the risk of myopathy. Use of simvastatin, lovastatin, or atorvastatin concomitantly with KETEK should be avoided. If KETEK is prescribed, therapy with simvastatin, lovastatin, or atorvastatin should be suspended during the course of treatment.
Monitoring of digoxin side effects or serum levels should be considered during concomitant administration of digoxin and KETEK. (See CLINICAL PHARMACOLOGY, Drug-drug interactions.)
Patients should be monitored with concomitant administration of midazolam and dosage adjustment of midazolam should be considered if necessary. Precaution should be used with other benzodiazepines, which are metabolized by CYP 3A4 and undergo a high first-pass effect (e.g., triazolam). (See CLINICAL PHARMACOLOGY, Drug-drug interactions.)
Concomitant treatment of KETEK with rifampin, a CYP 3A4 inducer, should be avoided. Concomitant administration of other CYP 3A4 inducers such as phenytoin, carbamazepine, or phenobarbital is likely to result in subtherapeutic levels of telithromycin and loss of effect. (See CLINICAL PHARMACOLOGY, Other drug interactions.)
In patients treated with metoprolol for heart failure, the increased exposure to metoprolol, a CYP 2D6 substrate, may be of clinical importance. Therefore, co-administration of KETEK and metoprolol in patients with heart failure should be considered with caution. (See CLINICAL PHARMACOLOGY, Drug-drug interactions.)
Spontaneous post-marketing reports suggest that administration of KETEK and oral anticoagulants concomitantly may potentiate the effectsof the oral anticoagulants. Consideration should be given to monitoring prothrombin times/INR while patients are receiving KETEK and oral anticoagulants simultaneously.
No specific drug interaction studies have been performed to evaluate the following potential drug-drug interactions with KETEK. However, these drug interactions have been observed with macrolide products.
-
Drugs metabolized by the cytochrome P450 system such as carbamazepine, cyclosporine, tacrolimus, sirolimus, hexobarbital, and phenytoin: elevation of serum levels of these drugs may be observed when co-administered with telithromycin. As a result, increases or prolongation of the therapeutic and/or adverse effects of the concomitant drug may be observed.
-
Ergot alkaloid derivatives (such as ergotamine or dihydroergotamine): acute ergot toxicity characterized by severe peripheral vasospasm and dysesthesia has been reported when macrolide antibiotics were co-administered. Without further data, the co-administration of KETEK and these drugs is not recommended.
Laboratory test interactions
There are no reported laboratory test interactions.
Carcinogenesis, mutagenesis, impairment of fertility
Long-term studies in animals to determine the carcinogenic potential of KETEK have not been conducted.
Telithromycin showed no evidence of genotoxicity in four tests: gene mutation in bacterial cells, gene mutation in mammalian cells, chromosome aberration in human lymphocytes, and the micronucleus test in the mouse.
No evidence of impaired fertility in the rat was observed at doses estimated to be 0.61 times the human daily dose on a mg/m2 basis. At doses of 1.8–3.6 times the human daily dose, at which signs of parental toxicity were observed, moderate reductions in fertility indices were noted in male and female animals treated with telithromycin.
Pregnancy
Teratogenic effects
Pregnancy Category C.
Telithromycin was not teratogenic in the rat or rabbit. Reproduction studies have been performed in rats and rabbits, with effect on pre-post natal development studied in the rat. At doses estimated to be 1.8 times (900 mg/m2) and 0.49 times (240 mg/m2) the daily human dose of 800 mg (492 mg/m2) in the rat and rabbit, respectively, no evidence of fetal terata was found. At doses higher than the 900 mg/m2 and 240 mg/m2 in rats and rabbits, respectively, maternal toxicity may have resulted in delayed fetal maturation. No adverse effects on prenatal and postnatal development of rat pups were observed at 1.5 times (750 mg/m2/d) the daily human dose.
There are no adequate and well-controlled studies in pregnant women. Telithromycin should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing mothers
Telithromycin is excreted in breast milk of rats. Telithromycin may also be excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when KETEK is administered to a nursing mother.
Pediatric use
The safety and effectiveness of KETEK in pediatric patients has not been established.
Geriatric use
In all Phase III clinical trials (n=4,780), KETEK was administered to 694 patients who were 65 years and older, including 231 patients who were 75 years and older. Efficacy and safety in elderly patients ≥ 65 years were generally similar to that observed in younger patients; however, greater sensitivity of some older individuals cannot be ruled out. No dosage adjustment is required based on age alone. (See CLINICAL PHARMACOLOGY, Special populations, Geriatric and DOSAGE AND ADMINISTRATION.)
ADVERSE REACTIONS
In Phase III clinical trials, 4,780 patients (n=2702 in controlled trials) received daily oral doses of KETEK 800 mg once daily for 5 days or 7 to 10 days. Most adverse events were mild to moderate in severity. In the combined Phase III studies, discontinuation due to treatment-emergent adverse events occurred in 4.4% of KETEK-treated patients and 4.3% of combined comparator-treated patients. Most discontinuations in the KETEK group were due to treatment-emergent adverse events in the gastrointestinal body system, primarily diarrhea (0.9% for KETEK vs. 0.7% for comparators), nausea (0.7% for KETEK vs. 0.5% for comparators).
All and possibly related treatment-emergent adverse events (TEAEs) occurring in controlled clinical studies in ≥ 2.0% of all patients are included below:
| All and Possibly Related Treatment-Emergent Adverse Events Reported in Controlled Phase III Clinical Studies (Percent Incidence) | ||||
|---|---|---|---|---|
| Adverse Event* | All TEAEs | Possibly-Related TEAEs | ||
| KETEK n=2702 |
Comparator† n=2139 |
KETEK n=2702 |
Comparator† n=2139 |
|
| Diarrhea | 10.8% | 8.6% | 10.0% | 8.0% |
| Nausea | 7.9% | 4.6% | 7.0% | 4.1% |
| Headache | 5.5% | 5.8% | 2.0% | 2.5% |
| Dizziness (excl. vertigo) | 3.7% | 2.7% | 2.8% | 1.5% |
| Vomiting | 2.9% | 2.2% | 2.4% | 1.4% |
| Loose Stools | 2.3% | 1.5% | 2.1% | 1.4% |
| Dysgeusia | 1.6% | 3.6% | 1.5% | 3.6% |
The following events judged by investigators to be at least possibly drug related were observed infrequently (≥ 0.2% and < 2%), in KETEK-treated patients in the controlled Phase III studies.
Gastrointestinal system: abdominal distension, dyspepsia, gastrointestinal upset, flatulence, constipation, gastroenteritis, gastritis, anorexia, oral candidiasis, glossitis, stomatitis, watery stools.
Liver and biliary system: abnormal liver function tests: increased transaminases, increased liver enzymes (e.g., ALT, AST) were usually asymptomatic and reversible. ALT elevations above 3 times the upper limit of normal were observed in 1.6%, and 1.7% of patients treated with KETEK and comparators, respectively. Hepatitis, with or without jaundice, occurred in 0.07% of patients treated with KETEK, and was reversible. (See PRECAUTIONS, General.)
Nervous system: dry mouth, somnolence, insomnia, vertigo, increased sweating
Body as a whole: abdominal pain, upper abdominal pain, fatigue
Special senses: Visual adverse events most often included blurred vision, diplopia, or difficulty focusing. Most events were mild to moderate; however, severe cases have been reported. Some patients discontinued therapy due to these adverse events. Visual adverse events were reported as having occurred after any dose during treatment, but most visual adverse events (65%) occurred following the first or second dose. Visual events lasted several hours and recurred upon subsequent dosing in some patients. For patients who continued treatment, some resolved on therapy while others continued to have symptoms until they completed the full course of treatment. (See PRECAUTIONS, General and PRECAUTIONS, Information for patients.)
Females and patients under 40 years old experienced a higher incidence of telithromycin-associated visual adverse events. (See CLINICAL STUDIES.)
Urogenital system: vaginal candidiasis, vaginitis, vaginosis fungal
Skin: rash
Hematologic: increased platelet count
Other possibly related clinically-relevant events occurring in <0.2% of patients treated with KETEK from the controlled Phase III studies included: anxiety, bradycardia, eczema, elevated blood bilirubin, erythema multiforme, flushing, hypotension, increased blood alkaline phosphatase, increased eosinophil count, paresthesia, pruritus, urticaria.
Post-Marketing Adverse Event Reports
In addition to adverse events reported from clinical trials, the following events have been reported from worldwide post-marketing experience with KETEK.
Allergic: face edema, rare reports of severe allergic reactions, including angioedema and anaphylaxis.
Cardiovascular: atrial arrhythmias, palpitations
Gastrointestinal system: pancreatitis
Liver and biliary system: Hepatic dysfunction has been reported. Severe and in some cases fatal hepatotoxicity, including fulminant hepatitis, hepatic necrosis and hepatic failure have been reported in patients treated with KETEK. These hepatic reactions were observed during or immediately after treatment. In some of these cases, liver injury progressed rapidly and occurred after administration of only a few doses of KETEK. (See CONTRAINDICATIONS and WARNINGS). Severe reactions, in some but not all cases, have been associated with serious underlying diseases or concomitant medications.
Data from post-marketing reports and clinical trials show that most reported cases of hepatic dysfunction were mild to moderate. (See PRECAUTIONS, General.)
Musculoskeletal: muscle cramps, rare reports of exacerbation of myasthenia gravis. (See WARNINGS.)
Nervous system: syncope usually associated with vagal syndrome.
OVERDOSAGE
In the event of acute overdosage, the stomach should be emptied by gastric lavage. The patient should be carefully monitored (e.g., ECG, electrolytes) and given symptomatic and supportive treatment. Adequate hydration should be maintained. The effectiveness of hemodialysis in an overdose situation with KETEK is unknown.
DOSAGE AND ADMINISTRATION
The dose of KETEK tablets is 800 mg taken orally once every 24 hours. The duration of therapy depends on the infection type and is described below. KETEK tablets can be administered with or without food.
| Infection | Daily dose and route of administration |
Frequency of administration | Duration of treatment |
|---|---|---|---|
| Acute bacterial exacerbation of chronic bronchitis | 800 mg oral (2 tablets of 400 mg) |
once daily | 5 days |
| Acute bacterial sinusitis | 800 mg oral (2 tablets of 400 mg) |
once daily | 5 days |
| Community-acquired pneumonia | 800 mg oral (2 tablets of 400 mg) |
once daily | 7–10 days |
KETEK may be administered without dosage adjustment in the presence of hepatic impairment.
In the presence of severe renal impairment (CLCR< 30 mL/min), including patients who need dialysis, the dose should be reduced to KETEK 600 mg once daily. In patients undergoing hemodialysis, KETEK should be given after the dialysis session on dialysis days. (See CLINICAL PHARMACOLOGY, Renal insufficiency.)
In the presence of severe renal impairment (CLCR< 30 mL/min), with coexisting hepatic impairment, the dose should be reduced to KETEK 400 mg once daily. (See CLINICAL PHARMACOLOGY, Multiple insufficiency.)
HOW SUPPLIED
KETEK® 400 mg tablets are supplied as light-orange, oval, film-coated tablets, imprinted "H3647" on one side and "400" on the other side. These are packaged in bottles and blister cards (Ketek Pak™ and unit dose) as follows:
Bottles of 60 (NDC 0088-2225-41)
Ketek Pak™, 10-tablet cards (2 tablets per blister cavity) (NDC 0088-2225-07)
Unit dose package of 100 (blister pack) (NDC 0088-2225-49)
KETEK® 300 mg tablets are supplied as light-orange, oval, film-coated tablets, imprinted "38AV" on one side and blank on the other side. These are packaged in bottles as follows:
Bottles of 20 (NDC 0088-2223-20)
Store at 25°C (77°F); excursions permitted to 15–30°C (59–86°F) [see USP Controlled Room Temperature].
CLINICAL STUDIES
Community-acquired pneumonia (CAP)
KETEK was studied in four randomized, double-blind, controlled studies and four open-label studies for the treatment of community-acquired pneumonia. Patients with mild to moderate CAP who were considered appropriate for oral outpatient treatment were enrolled in these trials. Patients with severe pneumonia were excluded based on any one of the following: ICU admission, need for parenteral antibiotics, respiratory rate > 30/minute, hypotension, altered mental status, < 90% oxygen saturation by pulse oximetry, or white blood cell count < 4000/mm3. Total number of clinically evaluable patients in the telithromycin group included 2016 patients.
| Patients (n) | Clinical cure rate | |||
|---|---|---|---|---|
| Controlled Studies | KETEK | Comparator | KETEK | Comparator |
|
||||
| KETEK vs. clarithromycin 500 mg BID for 10 days | 162 | 156 | 88.3% | 88.5% |
| KETEK vs. trovafloxacin* 200 mg QD for 7 to 10 days | 80 | 86 | 90.0% | 94.2% |
| KETEK vs. amoxicillin 1000 mg TID for 10 days | 149 | 152 | 94.6% | 90.1% |
| KETEK for 7 days vs. clarithromycin 500 mg BID for 10 days | 161 | 146 | 88.8% | 91.8% |
Clinical cure rates by pathogen from the four CAP controlled clinical trials in microbiologically evaluable patients given KETEK for 7–10 days or a comparator are displayed in Table 8.
| Pathogen | KETEK | Comparator |
|---|---|---|
| Streptococcus pneumoniae | 73/78 (93.6%) | 63/70 (90.0%) |
| Haemophilus influenzae | 39/47 (83.0%) | 42/44 (95.5%) |
| Moraxella catarrhalis | 12/14 (85.7%) | 7/9 (77.8%) |
| Chlamydophila (Chlamydia) pneumoniae | 23/25 (92.0%) | 18/19 (94.7%) |
| Mycoplasma pneumoniae | 22/23 (95.7%) | 20/22 (90.9%) |
Clinical cure rates for patients with CAP due to Streptococcus pneumoniae were determined from patients in controlled and uncontrolled trials. Of 333 evaluable patients with CAP due to Streptococcus pneumoniae, 312 (93.7%) achieved clinical success. Only patients considered appropriate for oral outpatient therapy were included in these trials. More severely ill patients were not enrolled. Blood cultures were obtained in all patients participating in the clinical trials of mild to moderate community-acquired pneumonia. In a limited number of outpatients with incidental pneumococcal bacteremia treated with KETEK, a clinical cure rate of 88% (67/76) has been observed. KETEK is not indicated for the treatment of severe community-acquired pneumonia or suspected pneumococcal bacteremia.
Clinical cure rates for patients with CAP due to multi-drug resistant Streptococcus pneumoniae (MDRSP*) were determined from patients in controlled and uncontrolled trials. Of 36 evaluable patients with CAP due to MDRSP, 33 (91.7%) achieved clinical success.
*MDRSP: Multi-drug resistant Streptococcus pneumoniae includes isolates known as PRSP (penicillin-resistant Streptococcus pneumoniae), and are isolates resistant to two or more of the following antibiotics: penicillin, 2nd generation cephalosporins, e.g., cefuroxime, macrolides, tetracyclines and trimethoprim/sulfamethoxazole.
| Screening Susceptibility | Clinical Success in Evaluable MDRSP Patients | |
|---|---|---|
| n/N* | % | |
| Penicillin-resistant | 20/23 | 86.9 |
| 2nd generation cephalosporin-resistant | 20/22 | 90.9 |
| Macrolide-resistant | 25/28 | 89.3 |
| Trimethoprim/sulfamethoxazole-resistant | 24/27 | 88.9 |
| Tetracycline-resistant† | 11/13 | 84.6 |
Acute bacterial sinusitis
KETEK was studied in two randomized, double-blind, comparative studies for the treatment of acute sinusitis. Clinical cure rates with KETEK given for 5 days and comparator drug are shown in Table 10.
| Patients (n) | Clinical cure rate | |||
|---|---|---|---|---|
| Controlled Studies | KETEK (5 day treatment) |
Comparator (10 day treatment) |
KETEK (5 day treatment) |
Comparator (10 day treatment) |
| KETEK vs. amoxicillin/clavulanic acid 500/125 mg TID | 146 | 137 | 75.3% | 74.5% |
| KETEK vs. cefuroxime axetil 250 mg BID | 189 | 89 | 85.2% | 82.0% |
A third study compared 5 days with 10 days of KETEK for the treatment of acute bacterial sinusitis, clinical cure rates for the two treatments were similar (91.1% vs. 91.0% respectively).
Clinical cure rates in microbiologically evaluable patients for KETEK against the most common pathogens from the two acute sinusitis controlled clinical trials are displayed in Table 11.
| Pathogen | KETEK 5 days | Comparator 10-days |
|---|---|---|
| Streptococcus pneumoniae | 27/31 (87.1%) | 14/16 (87.5%) |
| Haemophilus influenzae | 28/34 (82.4%) | 13/15 (86.7%) |
| Moraxella catarrhalis | 7/7 (100%) | 7/7 (100%) |
| Staphylococcus aureus | 8/8 (100%) | 2/3 (66.7%) |
Acute bacterial exacerbation of chronic bronchitis (AECB)
KETEK was studied in three randomized, double-blind, controlled studies for the treatment of acute exacerbation of chronic bronchitis. Clinical cure rates are displayed in Table 12.
| Controlled Studies | Patients (n) | Clinical cure rate | ||
|---|---|---|---|---|
| KETEK | Comparator | KETEK | Comparator | |
| KETEK (5 day therapy) vs. cefuroxime axetil 500mg BID (10 day therapy) | 140 | 142 | 86.4% | 83.1% |
| KETEK (5 day therapy) vs. amoxicillin/clavulanic acid 500/125 mg TID (10 day therapy) | 115 | 112 | 86.1% | 82.1% |
| KETEK (5 day therapy) vs. clarithromycin 500mg BID (10 day therapy) | 225 | 231 | 85.8% | 89.2% |
Clinical cure rates in microbiologically evaluable patients treated with KETEK against the most common pathogens from the three acute exacerbation of chronic bronchitis clinical trials are displayed in Table 13.
| Pathogen | KETEK | Comparator |
|---|---|---|
| Streptococcus pneumoniae | 22/27 (81.5%) | 15/19 (78.9%) |
| Haemophilus influenzae | 44/60 (73.3%) | 45/53 (84.9%) |
| Moraxella catarrhalis | 27/29 (93.1%) | 29/34 (85.3%) |
Visual Adverse Events
Table 14 provides the incidence of all treatment-emergent visual adverse events in controlled Phase III studies by age and gender. The group with the highest incidence was females under the age of 40, while males over the age of 40 had rates of visual adverse events similar to comparator-treated patients.
| Gender/Age | Telithromycin | Comparators* |
|---|---|---|
|
||
| Female ≤ 40 |
2.1% (14/682) |
0.0% (0/534) |
| Female > 40 |
1.0% (7/703) |
0.35% (2/574) |
| Male ≤ 40 |
1.2% (7/563) |
0.48% (2/417) |
| Male > 40 |
0.27% (2/754) |
0.33% (2/614) |
| Total | 1.1% (30/2702) |
0.28% (6/2139) |
ANIMAL PHARMACOLOGY
Repeated dose toxicity studies of 1, 3, and 6 months' duration with telithromycin conducted in rat, dog and monkey showed that the liver was the principal target for toxicity with elevations of liver enzymes and histological evidence of damage. There was evidence of reversibility after cessation of treatment. Plasma exposures based on free fraction of drug at the no observed adverse effect levels ranged from 1 to 10 times the expected clinical exposure.
Phospholipidosis (intracellular phospholipid accumulation) affecting a number of organs and tissues (e.g., liver, kidney, lung, thymus, spleen, gall bladder, mesenteric lymph nodes, GI-tract) has been observed with the administration of telithromycin in rats at repeated doses of 900 mg/m2/day (1.8× the human dose) or more for 1 month, and 300 mg/m2/day (0.61× the human dose) or more for 3–6 months. Similarly, phospholipidosis has been observed in dogs with telithromycin at repeated doses of 3000 mg/m2/day (6.1× the human dose) or more for 1 month and 1000 mg/m2/day (2.0× the human dose) or more for 3 months. The significance of these findings for humans is unknown.
Pharmacology/toxicology studies showed an effect both in prolonging QTc interval in dogs in vivo and in vitro action potential duration (APD) in rabbit Purkinje fibers. These effects were observed at concentrations of free drug at least 8.8 (in dogs) times those circulating in clinical use. In vitro electrophysiological studies (hERG assays) suggested an inhibition of the rapid activating component of the delayed rectifier potassium current (IKr) as an underlying mechanism.
Rev. June 2006
sanofi-aventis U.S. LLC
Bridgewater,
NJ 08807
© 2006 sanofi-aventis U.S. LLC
REFERENCES
- National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically – Sixth Edition; Approved Standard, NCCLS Document M7-A6, Vol. 23, No. 2, NCCLS, Wayne, PA, January, 2003.
- National Committee for Clinical Laboratory Standards. Performance Standards for Antimicrobial Disk Susceptibility Tests ‑ Eighth Edition; Approved Standard, NCCLS Document M2‑A8, Vol. 23, No. 1, NCCLS, Wayne, PA, January, 2003.
- National Committee for Clinical Laboratory Standards. Performance Standards for Antimicrobial Susceptibility Testing: Twelfth Informational Supplement; Approved Standard, NCCLS Document M2‑A8 and M7-A6, Vol. 23, No. 1, NCCLS, Wayne, PA, January, 2004.
PATIENT INFORMATION ABOUT:
KETEK®
(telithromycin)
Before beginning your treatment, please read this section to learn important information about KETEK® (telithromycin). Although the information presented here will be useful during your therapy, not all the benefits and risks of treatment with KETEK are discussed in this document. This section is not intended to take the place of conversations with your doctor or healthcare provider about your treatment or medical condition. The medicine described here can only be prescribed by a licensed healthcare provider. With this in mind, be sure to talk to your healthcare provider if you have any questions. It's important to note that only a doctor or healthcare provider can determine if KETEK is right for you.
What is KETEK?
KETEK (KEE tek) is an antibiotic used to treat adults 18 years of age and older with certain respiratory (lung and sinus) infections caused by certain germs called bacteria. KETEK kills many of the types of bacteria that can infect the lungs and sinuses, and has been found to treat these infections safely and effectively in clinical trials.
Not all respiratory infections are caused by bacteria. For example, common colds are caused by viruses. KETEK, like other antibiotics, does not kill viruses.
KETEK Tablets are light orange, oval, film-coated tablets each containing 400 mg or 300 mg of the active drug. The 400 mg tablet is imprinted with "H3647" on one side and "400" on the other side.
The 300 mg tablet is imprinted with "38AV" on one side and is blank on the other side.
How and when should I take KETEK?
The usual dose is two 400 mg KETEK Tablets taken at the same time once daily for 5 to 10 days. If you have kidney disease, with or without liver disease, your healthcare provider may change the dose prescribed for you.
KETEK tablets should be swallowed whole and may be taken with or without food. Try to take your tablets at the same time every day, unless your healthcare provider tells you otherwise.
Follow the dosing instructions carefully, and do not take more than the prescribed amount. If you miss a dose, take it as soon as you remember. Do not take more than one dose (e.g., two tablets) of KETEK in a 24-hour period. If you have any questions, talk to your healthcare provider.
To make sure that all bacteria are killed, take all of the medicine that was prescribed for you even if you begin to feel better, unless instructed otherwise. You should contact your healthcare provider if your condition is not improving while taking KETEK.
Who should not take KETEK?
You must not take KETEK if:
- You have ever had a severe allergic reaction to KETEK or to any of the group of antibiotics known as "macrolides" such as erythromycin, azithromycin (Zithromax®), clarithromycin (Biaxin®) or dirithromycin (Dynabac®).
- You are currently taking cisapride (Propulsid®) or pimozide (Orap®).
- You have ever experienced side effects on the liver while taking KETEK.
You should be sure to talk to your healthcare provider before taking KETEK if any of the following are true, so he/she can determine if KETEK is right for you:
- If you have, or if a relative has, a rare heart condition known as congenital prolongation of the QT interval.
- If you are being treated for heart rhythm disturbances with certain medicines known as antiarrhythmics (such as quinidine, procainamide, or dofetilide) or if you have low blood potassium (hypokalemia), or low blood magnesium (hypomagnesemia).
- If you have a disease known as myasthenia gravis.
- If you are pregnant, planning to become pregnant, or are nursing.
- If you have any other serious medical conditions, including heart, liver, or kidney disease.
What about other medications I am taking?
It is important to let your healthcare provider know about all of the medicines you are taking, including those obtained without a prescription. Also see section "Who should not take KETEK?"
It is important to tell your healthcare provider if you are taking:
- Simvastatin, lovastatin, or atorvastatin (used for lowering cholesterol). You should stop treatment with these medications while you are taking KETEK.
- Medicines that correct heart rhythm called "antiarrhythmics" (such as quinidine, procainamide, or dofetilide).
- Any of the following medicines: itraconazole, ketoconazole, midazolam, digoxin, ergot alkaloid derivatives, cyclosporine, carbamazepine, hexobarbital, phenytoin, tacrolimus, sirolimus, metoprolol, theophylline, rifampin or warfarin and other oral anticoagulants (sometimes called blood thinners).
- Medicines called diuretics (also sometimes called water pills) such as furosemide or hydrochlorothiazide.
What are the possible side effects of KETEK?
KETEK is generally well tolerated. Most side effects are mild to moderate.
The most common side effects are nausea, headache, dizziness, vomiting, and diarrhea. If diarrhea persists call your healthcare provider.
There have been reports of side effects on the liver, including severe liver disease. In some cases, liver damage worsened rapidly and happened after just a few doses of KETEK. If you develop signs or symptoms of hepatitis (liver disease), such as tiredness, body aches, loss of appetite, nausea, jaundice (yellow color of the skin and/or eyes), dark urine, light-colored stools, itchy skin, or belly pains, stop your medication and immediately contact your healthcare provider.
Worsening of myasthenia gravis has been reported in patients treated with KETEK. This has sometimes occurred within a few hours after taking the first dose . Reports have included death and life-threatening breathing trouble that happens fast in myasthenia gravis patients. If you have myasthenia gravis, you should talk with your doctor before taking KETEK.
KETEK may cause problems with vision, particularly when looking quickly between objects close by and objects far away. These events include blurred vision, difficulty focusing, and objects looking doubled. Most events were mild to moderate; however, severe cases have been reported. Problems with vision were reported as having occurred after any dose during treatment, but most occurred following the first or second dose. These problems lasted several hours and sometimes came back with the next dose.
If visual difficulties occur:
- You should avoid driving a motor vehicle, operating heavy machinery, or engaging in otherwise hazardous activities.
- Avoiding quickly looking between objects in the distance and objects nearby may help you to decrease these visual difficulties.
- You should contact your physician if these visual difficulties interfere with your daily activities.
You should be aware of the possibility of experiencing syncope (fainting), and its impact on the ability to drive, especially if you are experiencing vagal symptoms (severe nausea, vomiting, and/or lightheadedness). If you experience these symptoms, you should avoid driving a motor vehicle, operating heavy machinery, or engaging in otherwise hazardous activities.
KETEK has the potential to affect the heart, as seen on an electrocardiogram (EKG) test. In very rare cases, this condition may result in a serious abnormal heartbeat. Contact your healthcare provider if you have a fainting spell.
If you have other side effects not mentioned in this section or have concerns about side effects, be sure to talk to your healthcare provider.
How can I find out more about KETEK?
This is a summary of selected key points about KETEK. If you'd like more information or if you have concerns, talk to your healthcare provider. You can also visit the KETEK website at www.KETEK.com. But remember, neither this Patient Information nor the website can replace discussions with your doctor or healthcare provider.
Other key points to remember:
- Take your prescribed dose of KETEK once a day at the same time each day.
- Complete the course of medication (take all the tablets prescribed), even if you start to feel better, unless instructed otherwise.
- As with all other medications, do not use KETEK for other conditions or give tablets to others.
- Store KETEK tablets at room temperature.
- Keep this medication out of the reach of children.
- Do not take your tablets after the expiration date noted.
- Talk to your healthcare provider if you have questions or concerns.
Patient Information as of June 2006
BIAXIN®(clarithromycin) is a registered trademark of Abbott
Laboratories.
ZITHROMAX® (azithromycin) is a registered trademark of Pfizer Inc.
DYNABAC® (dirithromycin) is a registered trademark of Eli Lilly
and Company.
PROPULSID® (cisapride) is a registered trademark of Johnson & Johnson.
ORAP® (pimozide)
is a registered trademark of Teva Pharmaceuticals USA, Inc.
sanofi-aventis U.S. LLC
Bridgewater,
NJ 08807
© 2006 sanofi-aventis U.S. LLC
| Ketek (telithromycin) | ||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Ketek (telithromycin) | ||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
Revised: 06/2006Sanofi-Aventis U.S. LLC