LUMIZYME
-
alglucosidase alfa injection, powder, for solution
Genzyme Corporation
----------
|
||||||||||||||||||||
FULL PRESCRIBING INFORMATION
WARNING
Life-threatening anaphylactic
reactions, severe allergic reactions and immune mediated
reactions have been observed in some patients during
LUMIZYME™ infusions. Therefore, appropriate medical
support should be readily available when LUMIZYME is
administered [see Warnings and Precautions
(5.1,
5.2)].
Because of
the potential risk of rapid disease progression in Pompe disease
patients less than 8 years of age, LUMIZYME is available only
through a restricted distribution program called the LUMIZYME
ACE ProgramSM. Only prescribers and healthcare
facilities enrolled in the program may prescribe, dispense or
administer LUMIZYME. LUMIZYME may be administered only to
patients who are enrolled in and meet all the conditions of the
LUMIZYME ACE Program. To enroll in the LUMIZYME ACE Program call
1-800-745-4447 [see Warnings and Precautions
(5.3)].
1 INDICATIONS AND USAGE
LUMIZYME (alglucosidase alfa) [see Description (11)] is a lysosomal glycogen-specific enzyme indicated for patients 8 years and older with late (non-infantile) onset Pompe disease (acid α-glucosidase (GAA) deficiency) who do not have evidence of cardiac hypertrophy. The safety and efficacy of LUMIZYME have not been evaluated in controlled clinical trials in infantile-onset patients, or in late (non-infantile) onset patients less than 8 years of age [see Use in Specific Populations (8.4)].
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
The recommended dosage of LUMIZYME is 20 mg/kg body weight administered every 2 weeks as an intravenous infusion.
2.2 Instructions for Use
LUMIZYME does not contain any preservatives. Vials are
single-use only. Discard any unused product.
The
total volume of infusion is determined by the patient’s body
weight and should be administered over approximately 4 hours.
Infusions should be administered in a step-wise manner using an
infusion pump. The initial infusion rate should be no more than
1 mg/kg/hr. The infusion rate may be increased by 2 mg/kg/hr
every 30 minutes, after patient tolerance to the infusion rate
is established, until a maximum rate of 7 mg/kg/hr is reached.
Vital signs should be obtained at the end of each step. If the
patient is stable, LUMIZYME may be administered at the maximum
rate of 7 mg/kg/hr until the infusion is completed. The infusion
rate may be slowed or temporarily stopped in the event of
infusion reactions. See Table 1
below for the rate of infusion at each step, expressed as mL/hr
based on the recommended infusion volume by patient weight.
| Patient
Weight Range (kg) | Total
infusion volume (mL) | Step 1 1 mg/kg/hr (mL/hr) |
Step
2 |
Step 3 |
Step
4 |
|
20.1 – 30 | 150 | 8 | 23 | 38 | 53 |
|
30.1 – 35 | 200 | 10 | 30 | 50 | 70 |
| 35.1 – 50 | 250 | 13 | 38 | 63 | 88 |
| 50.1 – 60 | 300 | 15 | 45 | 75 | 105 |
| 60.1 – 100 | 500 | 25 | 75 | 125 | 175 |
| 100.1 – 120 | 600 | 30 | 90 | 150 | 210 |
| 120.1 – 140 | 700 | 35 | 105 | 175 | 245 |
| 140.1 – 160 | 800 | 40 | 120 | 200 | 280 |
| 160.1 – 180 | 900 | 45 | 135 | 225 | 315 |
| 180.1 – 200 | 1,000 | 50 | 150 | 250 | 350 |
2.3 Reconstitution, Dilution, and Administration
LUMIZYME should be reconstituted, diluted and administered by a health care professional.
Use aseptic technique during preparation. Do
not use filter needles during preparation.
- Determine the number of vials to be reconstituted based on
the individual patient’s weight and the
recommended dose of 20 mg/kg.
Patient weight (kg) x dose (mg/kg) = patient dose (in mg)
Patient dose (in mg) divided by 50 mg/vial = number of vials to reconstitute. If the number of vials includes a fraction, round up to the next whole number.
Example: Patient weight (68 kg) x dose (20 mg/kg) = patient dose (1,360 mg)
1,360 mg divided by 50 mg/vial = 27.2 vials; therefore, 28 vials should be reconstituted
Remove the required number of vials from the refrigerator and allow them to reach room temperature prior to reconstitution (approximately 30 minutes).
- Reconstitute each LUMIZYME vial by slowly injecting 10.3
mL of Sterile Water for Injection, USP to the inside wall of
each vial. Each vial will yield a concentration of 5 mg/mL.
The total extractable dose per vial is 50 mg per 10 mL.
Avoid forceful impact of the water for injection on the
powder and avoid foaming. This is done by slow drop-wise
addition of the water for injection down the inside of the
vial and not directly onto the lyophilized cake. Tilt and
roll each vial gently. Do not invert, swirl, or
shake.
- The reconstituted LUMIZYME solution should be protected
from light.
- Perform an immediate visual inspection on the
reconstituted vials for particulate matter and
discoloration. If upon immediate inspection opaque particles
are observed or if the solution is discolored do not use.
The reconstituted solution may occasionally contain some
alglucosidase alfa particles (typically less than 10 in a
vial) in the form of thin white strands or translucent
fibers subsequent to the initial inspection. This may also
happen following dilution for infusion. These particles have
been shown to contain alglucosidase alfa and may appear
after the initial reconstitution step and increase over
time. Studies have shown that these particles are removed
via in-line filtration without having a detectable effect on
the purity or strength.
- LUMIZYME should be diluted in 0.9% Sodium Chloride
for Injection, USP, immediately after reconstitution, to a
final LUMIZYME concentration of 0.5 to 4 mg/mL.
See Table
1 for the recommended total infusion volume based
on patient weight.
- Slowly withdraw the reconstituted solution from each
vial. Avoid foaming in the
syringe.
- Remove airspace from the infusion bag to minimize particle
formation due to the sensitivity of LUMIZYME to air-liquid
interfaces.
- Add the reconstituted LUMIZYME solution slowly and
directly into the sodium chloride solution. Do not
add directly into airspace that may remain within the
infusion bag. Avoid foaming in the infusion
bag.
- Gently invert or massage the infusion bag to
mix. Do not shake.
- Administer LUMIZYME using an in-line low protein binding
0.2 µm filter.
- Do not infuse LUMIZYME in the same intravenous line with other products.
LUMIZYME does not contain any preservatives. Vials are
single-use only.
Discard any unused product.
3 DOSAGE FORMS AND STRENGTHS
LUMIZYME is supplied as a sterile, nonpyrogenic, white to
off-white, lyophilized cake or powder for reconstitution with Sterile
Water for Injection, USP to yield a concentration of 5 mg/mL; and then
further diluted with 0.9% Sodium Chloride for Injection, USP for
intravenous infusion.
Single use vials are available in 50 mg
dosage only.
4 CONTRAINDICATIONS
None.
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis and Allergic Reactions
(see Boxed Warning)
Anaphylaxis and severe allergic reactions have been observed in patients during and up to 3 hours after LUMIZYME infusion. Some of the reactions were life-threatening and included anaphylactic shock, respiratory arrest, apnea, dyspnea, bradycardia, tachycardia, and hypotension. Other accompanying reactions included chest discomfort/pain, throat tightness, bronchospasm, wheezing, tachypnea, cyanosis, decreased oxygen saturation/hypoxia, convulsions, angioedema (including tongue or lip swelling, periorbital edema, and face edema), pruritus, rash, urticaria, hyperhidrosis, nausea, dizziness, hypertension, flushing/erythema, fever, pallor, peripheral coldness, feeling hot, restlessness, nervousness, headache, back pain, and paraesthesia. Some of these reactions were IgE-mediated [see Adverse Reactions (6.2)].
If anaphylaxis or other severe allergic reactions occur, immediate discontinuation of the administration of LUMIZYME should be considered, and appropriate medical treatment should be initiated. Severe reactions are generally managed with infusion interruption, administration of antihistamines, corticosteroids, intravenous fluids, and/or oxygen, when clinically indicated. In some cases of anaphylaxis, epinephrine has been administered. Because of the potential for severe allergic reactions, appropriate medical support, including cardiopulmonary resuscitation equipment, should be readily available when LUMIZYME is administered.
The risks and benefits of re-administering LUMIZYME following an anaphylactic or severe allergic reaction should be considered. Some patients have been rechallenged and have continued to receive LUMIZYME under close clinical supervision. Extreme care should be exercised, with appropriate resuscitation measures available, if the decision is made to re-administer the product [see Adverse Reactions (6.2)].
5.2 Immune Mediated Reactions
(see Boxed Warning)
Severe cutaneous reactions have been reported with alglucosidase alfa including necrotizing skin lesions [see Adverse Reactions (6.3)]. Systemic immune mediated reactions, including possible type III immune complex-mediated reactions have been observed with alglucosidase alfa. These reactions occurred several weeks to 3 years after initiation of alglucosidase alfa infusions. Skin biopsy in one patient demonstrated deposition of anti-rhGAA antibodies in the lesion. Another patient developed severe inflammatory arthropathy in association with fever and elevated erythrocyte sedimentation rate. Patients should be monitored for the development of systemic immune complex-mediated reactions involving skin and other organs while receiving LUMIZYME. If immune mediated reactions occur, discontinuation of the administration of LUMIZYME should be considered, and appropriate medical treatment initiated. The risks and benefits of re-administering alglucosidase alfa following an immune mediated reaction should be considered. Some patients have successfully been rechallenged and have continued to receive alglucosidase alfa under close clinical supervision.
5.3 Distribution Program for LUMIZYME
(see Boxed Warning)
Lumizyme is available only under a restricted distribution program called the Lumizyme ACE (Alglucosidase Alfa Control and Education) Program.
The purpose of the program is to ensure that the known risks of anaphylaxis and severe allergic reactions and the potential risks of severe cutaneous and systemic immune complex-mediated reactions associated with the use of LUMIZYME are communicated to patients, caregivers, and prescribers. In addition, the purpose of the program is to mitigate the potential risk of rapid disease progression in infantile-onset Pompe disease patients and late (non-infantile) onset Pompe disease patients less than 8 years of age for whom the safety and effectiveness of LUMIZYME have not been evaluated.
Under this program, only trained and certified prescribers, and healthcare facilities enrolled in the program are able to prescribe, dispense or administer Lumizyme, and only patients who are enrolled in and meet all the conditions of the Lumizyme ACE Program may receive Lumizyme.
For information about the ACE Program call 1-800-745-4447.
5.4 Risk of Acute Cardiorespiratory Failure
Patients with acute underlying respiratory illness or compromised cardiac and/or respiratory function may be at risk of serious exacerbation of their cardiac or respiratory compromise during infusions. Appropriate medical support and monitoring measures should be readily available during LUMIZYME infusion, and some patients may require prolonged observation times that should be based on the individual needs of the patient. Acute cardiorespiratory failure has been observed in a few infantile-onset Pompe disease patients with underlying cardiac hypertrophy, possibly associated with fluid overload with intravenous administration of alglucosidase alfa [see Dosage and Administration (2.2)].
5.5 Precautions for General/Regional Anesthesia
Administration of general anesthesia can be complicated by the presence of severe cardiac and skeletal (including respiratory) muscle weakness. Cardiac arrhythmia has been observed in infantile-onset patients with cardiac hypertrophy, associated with the use of general anesthesia. As Pompe disease is considered a neuromuscular disease, caution should be used when administering general anesthesia in Pompe disease patients.
5.6 Monitoring: Laboratory Tests
Patients should be monitored for IgG antibody formation every 3 months for 2 years and then annually thereafter. Testing for IgG titers may also be considered if patients develop allergic or other immune mediated reactions. Patients who experience anaphylactic or allergic reactions may also be tested for IgE antibodies to alglucosidase alfa and other mediators of anaphylaxis [see Adverse Reactions (6.2)].
There are currently no marketed tests for antibodies against alglucosidase alfa, however a testing service is provided by Genzyme. Contact your local Genzyme representative or Genzyme Corporation at 1-800-745-4447 for information on testing and to obtain a sample collection box.
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under controlled conditions, the observed adverse reaction rates may not predict the rates observed in patients in clinical practice. Assessment of adverse reactions is based on the exposure of 90 patients (45 male, 45 female) with late-onset Pompe disease, ages 10 to 70 years, to 20 mg/kg LUMIZYME or placebo in a randomized, double-blind, placebo-controlled study designed to enroll patients age 8-70 years. The youngest LUMIZYME-treated patient was 16 years of age, and the youngest placebo-treated patient was 10 years of age. All patients were naïve to enzyme replacement therapy. Patients were randomized in a 2:1 ratio and received LUMIZYME or placebo every other week for 78 weeks (18 months). The study population included 34 males and 26 females (N=60) in the LUMIZYME group and 11 males and 19 females (N=30) in the placebo group. Two patients receiving LUMIZYME discontinued the study due to anaphylactic reactions. A third patient in the LUMIZYME group died during the study due to brain stem ischemia secondary to thrombosis of a basilar aneurysm, which was considered unrelated to treatment.
Serious adverse reactions reported with LUMIZYME in the randomized, double-blind, placebo-controlled study included anaphylaxis [see Boxed Warning and Warnings and Precautions (5.1)]. Anaphylactic reactions included: angioedema, throat tightness and chest pain/discomfort. One patient with a history of Wolff-Parkinson-White syndrome experienced a serious adverse reaction of supraventricular tachycardia. Other serious adverse events that occurred in a higher incidence in LUMIZYME treated patients compared to placebo included coronary artery disease, intervertebral disc protrusion, pneumonia, gastroenteritis, and dehydration.
The most common adverse reactions observed were infusion reactions. Infusion reactions, defined as an adverse reaction occurring during the infusion or within 2 hours after completion of the infusion, that occurred in LUMIZYME treated patients at an incidence of ≥ 5% compared to placebo in the controlled study included anaphylaxis, urticaria, diarrhea, vomiting, dyspnea, pruritus, rash/erythema, pharyngolaryngeal pain, neck pain, hypoacusis, flushing/feeling hot, pain in extremity, fall and chest discomfort. Additional infusion reactions observed in other clinical trials and expanded access programs with LUMIZYME included respiratory distress, cough, livedo reticularis, agitation, irritability, retching, rigors, tremor and increased lacrimation.
If an infusion reaction occurs, decreasing the infusion rate, temporarily stopping the infusion, and/or administration of antihistamines and/or antipyretics may ameliorate the symptoms. If severe infusion or allergic reactions occur, immediate discontinuation of the administration of LUMIZYME should be considered, and appropriate medical treatment should be initiated [see Warnings and Precautions (5.1)]. Severe infusion reactions are generally managed with infusion interruption, administration of antihistamines, corticosteroids, intravenous fluids, and/or oxygen, when clinically indicated. In some cases of anaphylactic reactions, epinephrine was administered. Patients who have experienced infusion reactions should be treated with caution when they are re-administered LUMIZYME.
Delayed onset infusion reactions have also been observed with LUMIZYME infusion. Delayed onset infusion reactions, defined as adverse reactions that occurred within 48 hours after completion of LUMIZYME infusion, occurred in LUMIZYME treated patients at an incidence of ≥ 3% compared to placebo treated patients in a controlled trial. Symptoms included urticaria, dizziness, procedural pain, pharyngolaryngeal pain, malaise, muscle spasms, musculoskeletal pain, musculoskeletal weakness, musculoskeletal stiffness, neck pain, insomnia, and epistaxis. Patients should be counseled about the possibility of delayed onset infusion reactions and given proper follow up instructions.
Table 2 enumerates adverse reactions that occurred in LUMIZYME treated patients at an incidence of ≥ 5% compared to placebo treated patients during the randomized, double-blind, placebo-controlled study. Reported adverse reactions have been classified by Medical Dictionary for Regulatory Activities (MedDRA) terminology System Organ Class and Preferred Term.
|
System Organ Class |
Preferred Term |
LUMIZYME n=60 N (%) |
Placebo n=30 N (%) |
|
Blood and lymphatic system disorders |
Lymphadenopathy |
5 (8.3) |
0 (0) |
|
Ear and labyrinth disorders |
Hypoacusis |
20 (33.3) |
7 (23.3) |
|
Vertigo |
4 (6.7) |
0 (0) |
|
|
Ear discomfort or pain |
7 (11.7) |
2 (6.7) |
|
|
Eye disorders |
Vision blurred |
3 (5) |
0 (0) |
|
Gastrointestinal disorders |
Constipation |
6 (10) |
0 (0) |
|
Dyspepsia |
5 (8.3) |
0 (0) |
|
|
Vomiting |
13 (21.7) |
3 (10) |
|
|
General disorders and administration
site |
Chest discomfort or pain |
10 (16.7) |
2 (6.7) |
|
Infusion site reactions |
8 (13.3) |
0 (0) |
|
|
Malaise |
3 (5) |
0 (0) |
|
|
Edema, peripheral |
10 (16.7) |
3 (10) |
|
|
Pain |
5 (8.3) |
1 (3.3) |
|
|
Immune system disorders |
Anaphylaxis |
4 (6.7) |
0 (0) |
|
Infections and infestations |
Gastroenteritis |
6 (10) |
1 (3.3) |
|
Respiratory tract infection |
3 (5) |
0 (0) |
|
|
Upper respiratory tract infection |
11 (18.3) |
3 (10) |
|
|
Injury, poisoning and procedural complications |
Procedural pain |
9 (15) |
3 (10) |
|
Metabolism and nutrition disorders |
Hypokalemia |
3 (5) |
0 (0) |
|
Musculoskeletal and connective tissue disorders |
Muscle twitching |
5 (8.3) |
1 (3.3) |
|
Musculoskeletal pain |
22 (36.7) |
9 (30) |
|
|
Musculoskeletal stiffness or tightness |
9 (15) |
2 (6.7) |
|
|
Nervous system disorders |
Somnolence |
3 (5) |
0 (0) |
|
Tremor |
4 (6.7) |
0 (0) |
|
|
Renal and urinary disorders |
Nephrolithiasis |
3 (5) |
0 (0) |
|
Respiratory, thoracic and mediastinal disorders |
Dyspnea, exertional |
4 (6.7) |
0 (0) |
|
Epistaxis |
3 (5) |
0 (0) |
|
|
Skin and subcutaneous tissue disorders |
Hyperhidrosis |
5 (8.3) |
0 (0) |
|
Pruritis |
6 (10) |
1 (3.3) |
|
|
Urticaria |
6 (10) |
0 (0) |
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The data reflect the percentage of patients whose tests results were considered positive for antibodies to alglucosidase alfa using an enzyme-linked immunosorbent assay (ELISA) and confirmed by a radioimmunoprecipitation (RIP) assay for alglucosidase alfa-specific IgG antibodies. The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including inhibitory antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to alglucosidase alfa with the incidence of antibodies to other products may be misleading.
In the randomized, double-blind, placebo-controlled study, all patients with available samples treated with LUMIZYME (N=59, 100%) developed IgG antibodies to alglucosidase alfa. All patients who developed IgG antibodies did so within the first 3 months of exposure (median time to seroconversion was 4 weeks). There was no apparent association between mean or peak IgG antibody titers and the occurrence of adverse reactions.
Patients who developed IgG antibodies to alglucosidase alfa were also evaluated for inhibition of enzyme activity or cellular uptake of enzyme in in vitro assays. None of the 59 evaluable patients tested positive for inhibition of enzyme activity. Antibody titers for cellular uptake inhibition were present in 18 of 59 patients (31%) by Week 78. All other patients tested negative for inhibition of cellular uptake. Patients who were positive for uptake inhibition tended to have higher IgG titers than patients who tested negative for uptake inhibition. Among the 32 patients with evaluable pharmacokinetic (PK) samples, 5 patients tested positive for uptake inhibition at times corresponding to PK sampling times as compared to other patients. The clearance values for 4 of these 5 patients were approximately 1.2- to 1.8-fold greater in the presence (Week 52) as compared to in the absence of inhibitory antibodies (Week 0) [see Clinical Pharmacology (12.3)].
Patients in the clinical studies or in the postmarketing setting have undergone testing for alglucosidase alfa-specific IgE antibodies. Testing was performed in patients who experienced moderate to severe or recurrent infusion reactions, for which mast-cell activation was suspected.
Ten patients in the randomized, double-blind, placebo-controlled study underwent testing for alglucosidase alfa-specific IgE antibodies. Two of 10 patients evaluated tested positive for alglucosidase alfa-specific IgE-binding antibodies, both of whom experienced anaphylactic reactions [see Boxed Warning and Warnings and Precautions (5.1)]. One patient who developed IgE antibodies discontinued the study following anaphylaxis.
A small number of LUMIZYME treated patients in the postmarketing setting who were evaluated tested positive for presence of alglucosidase alfa-specific IgE antibodies. Some of these patients experienced anaphylaxis [see Boxed Warning and Warnings and Precautions (5.1)] .
Some patients who tested positive for alglucosidase alfa-specific IgE antibodies were successfully rechallenged with LUMIZYME using a slower infusion rate at lower initial doses and have continued to receive treatment under close clinical supervision [see Warnings and Precautions (5.1)].
Patients who develop IgE antibodies to alglucosidase alfa appear to be at a higher risk for the occurrence of anaphylaxis and severe allergic reactions [see Warnings and Precautions (5.1)]. Therefore, these patients should be monitored more closely during administration of LUMIZYME.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post approval use of LUMIZYME. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. In postmarketing experience with LUMIZYME, deaths, and serious adverse reactions have been reported, including anaphylaxis [see Boxed Warning and Warnings and Precautions (5.1)]. Adverse events resulting in death reported in the postmarketing setting with LUMIZYME treatment included cardiorespiratory arrest, respiratory failure, hemothorax, pneumothorax, cardiac failure, sepsis, aortic dissection, cerebrovascular accident, and skin necrosis. The most frequently reported serious adverse reactions were infusion reactions. In addition to the infusion reactions reported in clinical trials [see Adverse Reactions (6.1)], the following serious adverse events have been reported in at least 2 patients: dyspnea, respiratory failure, bronchospasm, stridor, decreased oxygen saturation/hypoxia, pharyngeal edema, chest discomfort, chest pain, hypotension, hypertension, erythema, flushing, lung infection, tachycardia, cyanosis, and hypersensitivity. One case of hyperparathyroidism has been reported.
Systemic and cutaneous immune mediated reactions, including necrotizing skin lesions have been reported in postmarketing safety experience with alglucosidase alfa [see Warnings and Precautions (5.2)].
7 DRUG INTERACTIONS
7.1 Interference with Other Drugs
No drug interaction or in vitro metabolism studies were performed.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Teratogenic Effects
Pregnancy Category B. Reproduction studies have been performed in pregnant mice at intravenous doses up to 40 mg/kg/day (plasma AUC of 64.6 mg·min/mL, 0.4 times the human steady-state exposure at the recommended bi-weekly dose) and pregnant rabbits at intravenous doses up to 40 mg/kg/day (plasma AUC of 85 mg·min/mL, 0.5 times the human steady-state exposure at the recommended bi-weekly dose) and have revealed no evidence of impaired fertility or harm to the fetus due to alglucosidase alfa. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Women of childbearing potential are encouraged to enroll in the Pompe Registry [see Patient Counseling Information (17)].
8.2 Labor and Delivery
Information on the effect of LUMIZYME on labor and delivery is unknown. Pregnant women are encouraged to enroll in the Pompe Registry [see Patient Counseling Information (17)].
8.3 Nursing Mothers
It is not known whether LUMIZYME is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when LUMIZYME is administered to a nursing woman. Nursing women are encouraged to enroll in the Pompe Registry [see Patient Counseling Information (17)].
8.4 Pediatric Use
LUMIZYME is not for use in patients with infantile-onset Pompe disease or late (non-infantile) onset Pompe disease who are less than 8 years of age. The safety and effectiveness of LUMIZYME in these patients have not been evaluated in clinical trials.
The safety and effectiveness of LUMIZYME was assessed in a randomized, double-blind, placebo-controlled study of 90 patients with late (non-infantile) onset Pompe disease. Patients age 8 to 70 years were eligible for enrollment. The study included 2 patients 16 years of age or less (n=1, age 16 years, LUMIZYME treatment group, n=1, age 10 years, placebo group) [see Clinical Studies (14.1)].
8.5 Geriatric Use
The randomized, double-blind, placebo-controlled study of LUMIZYME did not include sufficient numbers (n=4) of patients aged 65 years and over to determine whether they respond differently from younger patients [see Clinical Studies (14.1)].
10 OVERDOSAGE
There have been no reports of overdose with LUMIZYME. In the placebo-controlled study, patients received doses up to 20 mg/kg body weight every other week.
11 DESCRIPTION
LUMIZYME (alglucosidase alfa) consists of the human enzyme acid α-glucosidase (GAA), encoded by the most predominant of nine observed haplotypes of this gene. LUMIZYME is produced by recombinant DNA technology in a Chinese hamster ovary cell line. The LUMIZYME manufacturing process differs from that for MYOZYME, resulting in differences in some product attributes. Alglucosidase alfa degrades glycogen by catalyzing the hydrolysis of α-1,4- and α-1,6 glycosidic linkages of lysosomal glycogen.
Alglucosidase alfa is a glycoprotein with a calculated mass of 99,377 daltons for the polypeptide chain, and a total mass of approximately 109,000 daltons, including carbohydrates. Alglucosidase alfa has a specific activity of 3 to 5 Units/mg (one unit is defined as that amount of activity that results in the hydrolysis of 1 micro mole of synthetic substrate per minute under specified assay conditions). LUMIZYME is intended for intravenous infusion. It is supplied as a sterile, nonpyrogenic, white to off-white, lyophilized cake or powder for reconstitution with 10.3 mL Sterile Water for Injection, USP. Each 50 mg vial contains 52.5 mg alglucosidase alfa, 210 mg mannitol, 0.5 mg polysorbate 80, 9.9 mg sodium phosphate dibasic heptahydrate, 31.2 mg sodium phosphate monobasic monohydrate. Following reconstitution as directed, each vial contains 10.5 mL reconstituted solution and a total extractable volume of 10 mL at 5 mg/mL alglucosidase alfa. LUMIZYME does not contain preservatives; each vial is for single use only.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pompe disease (acid maltase deficiency, glycogen storage disease type II, GSD II, glycogenosis type II) is an inherited disorder of glycogen metabolism caused by the absence or marked deficiency of the lysosomal enzyme GAA.
LUMIZYME provides an exogenous source of GAA. Binding to mannose-6-phosphate receptors on the cell surface has been shown to occur via carbohydrate groups on the GAA molecule, after which it is internalized and transported into lysosomes, where it undergoes proteolytic cleavage that results in increased enzymatic activity. It then exerts enzymatic activity in cleaving glycogen.
12.2 Pharmacodynamics
Clinical pharmacodynamic studies have not been conducted for LUMIZYME.
12.3 Pharmacokinetics
The pharmacokinetics of alglucosidase alfa were studied in 32 late-onset Pompe disease patients from the randomized, double-blind, placebo-controlled study ranging in age from 21 to 70 years old who received LUMIZYME 20 mg/kg every other week. The pharmacokinetics were not-time dependent for patients who did not develop high antibody titer/inhibitory antibody. Parameter values did not change across visits at Weeks 0, 12, and 52. At Week 52 of bi-weekly administration the estimates of AUC (2700 mcg·h/mL with 30.4% coefficient of variation [CV],n=29), Cmax (372 mcg /mL with 22.7% CV,n=29) and clearance (601 mL/h with 28.2% CV,n=29) were determined at steady-state. The declining portion of the concentration-time profile of alglucosidase alfa appears biphasic within the observed sampling time. The half-life for the first phase is 2.4 hours with a between subject variation of 10%. Concentrations of alglucosidase alfa were not sampled long enough to adequately determine the half-life for the second phase.
Higher mean clearance (42%) was observed at Week 52 in 4 of 5 patients that tested positive for antibodies that inhibit the cellular uptake of enzyme. Pharmacokinetics in 4 of these 5 individuals over time indicated an increase in clearance with increase in IgG titer. Positive inhibitory antibody status correlated with higher IgG titers in patients who received LUMIZYME. The relationship between exposure and efficacy has not been defined.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to evaluate carcinogenic potential or studies to evaluate mutagenic potential have not been performed with alglucosidase alfa.
Alglucosidase alfa at intravenous doses up to 40 mg/kg, administered every other day (plasma AUC of 64.6 mg·min/mL, 0.4 times the human exposure at the recommended bi-weekly dose) had no effect on fertility and reproductive performance in mice.
14 CLINICAL STUDIES
14.1 Controlled clinical trials
The safety and efficacy of LUMIZYME was assessed in 90 patients with late-onset Pompe disease, ages 10 to 70 years, in a randomized double-blind, placebo-controlled study designed to enroll patients age 8-70 years. The youngest LUMIZYME-treated patient was 16 years of age, and the youngest placebo-treated patient was 10 years of age. All patients were naïve to enzyme replacement therapy. Patients were allocated in a 2:1 ratio and received 20 mg/kg LUMIZYME (n=60) or placebo (n=30) every other week for 78 weeks (18 months). The study population included 34 males and 26 females (N=60) in the LUMIZYME group and 11 males and 19 females (N=30) in the placebo group. At baseline, all patients were ambulatory (some required assistive walking devices), did not require invasive ventilator support or non-invasive ventilation while awake and sitting upright and had a forced vital capacity (FVC) between 30 and 79% of predicted in the sitting position. Patients who could not walk 40 meters in 6 minutes or were unable to perform appropriate pulmonary and muscle function testing were excluded from the study.
A total of 81 of 90 patients completed the study. Of the 9 patients who discontinued, 5 were in the LUMIZYME group and 4 were in the placebo group. Three patients discontinued the study due to an adverse event; two patients were in the LUMIZYME treatment group and one patient was in placebo group. One patient in the LUMIZYME group died [see Adverse Reactions (6.1)]. Four patients discontinued study participation to pursue treatment with commercial therapy, and one patient discontinued the study for personal reasons.
At study entry, the mean % predicted FVC in the sitting position among all patients was about 55%. After 78 weeks, the mean % predicted FVC increased to 56.2% for LUMIZYME-treated patients and decreased to 52.8% for placebo-treated patients indicating a LUMIZYME treatment effect of 3.4 % (95% confidence interval: [1.3% to 5.5%]; p=0.004). Stabilization of % predicted FVC in the LUMIZYME-treated patients was observed (see Figure 1).
Figure 1: Mean FVC Upright (% Predicted) Over Time
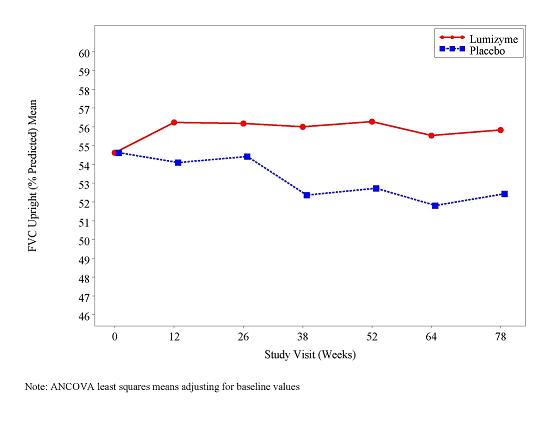
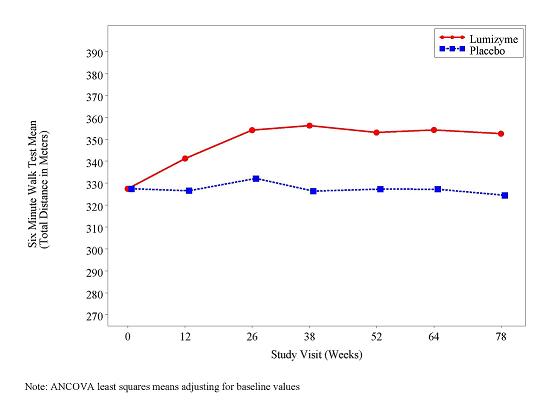
At study entry, the mean 6 minute walk test (6MWT) among all patients was about 330 meters. After 78 weeks, the mean 6MWT increased by 25 meters for LUMIZYME-treated patients and decreased by 3 meters for placebo-treated patients indicating a LUMIZYME treatment effect of 28 meters (95% confidence interval: [-1 to 52 meters]; p=0.06) (see Figure 2).
Figure 2: Mean Six Minute Walk Test Total Distance Walked Over Time
14.2 Uncontrolled studies
The effectiveness of Lumizyme has not been established in infantile-onset patients. Descriptive data from infantile-onset patients who have received Lumizyme commercially outside the U.S. have been collected in the Pompe Registry. The Pompe Registry is a multi-center, multi-national, voluntary, observational disease registry. Fifteen infantile-onset patients enrolled in the registry were matched to the baseline characteristics of an untreated historical control cohort. These patients were diagnosed with Pompe disease and received treatment with LUMIZYME prior to 6 months of age (range 0.6 to 6 months). The median duration of treatment was 15 months (range 3 to 48 months). Estimated survival in LUMIZYME-treated patients was 57% at 18 months and 37% at 36 months, compared to the 2% survival in the historical control group at both time points. The median age of death or last follow-up was 19 months (range 5 to 51 months).
Descriptive clinical data from patients with infantile-onset Pompe disease in the Pompe Registry were used to verify the overall effectiveness of LUMIZYME for patients 8 years and older with late-onset Pompe disease.
16 HOW SUPPLIED/STORAGE AND HANDLING
LUMIZYME 50 mg vials are supplied as a sterile, nonpyrogenic, white to off-white lyophilized cake or powder. LUMIZYME is supplied in single-use, clear Type I glass 20 mL (cc) vials. The closure consists of a siliconized butyl stopper and an aluminum seal with a plastic flip-off cap.
Store LUMIZYME under refrigeration between 2° to 8°C (36° to 46°F). Do not use LUMIZYME after the expiration date on the vial.
The reconstituted and diluted solution should be administered without delay. If immediate use is not possible, the reconstituted and diluted solution is stable for up to 24 hours at 2° to 8°C (36° to 46°F). Storage of the reconstituted solution at room temperature is not recommended. The reconstituted and diluted LUMIZYME solution should be protected from light. Do not freeze or shake.
LUMIZYME does not contain any preservatives. Vials are single-use only. Discard any unused product
NDC 58468-0160-1
17 PATIENT COUNSELING INFORMATION
17.1 Distribution Program for LUMIZYME
Patients and caregivers should be informed that Lumizyme is available only under a restricted distribution program called the LUMIZYME ACE (Alglucosidase Alfa Control and Education) Program.
The purpose of the program is to ensure that the known risks of anaphylaxis and severe allergic reactions and the potential risks of severe cutaneous and systemic immune complex-mediated reactions associated with the use of LUMIZYME are communicated to patients, caregivers, and prescribers. In addition, the purpose of the program is to mitigate the potential risk of rapid disease progression in infantile-onset Pompe disease patients and late (non-infantile) onset Pompe disease patients less than 8 years of age with for whom the safety and effectiveness of LUMIZYME have not been evaluated.
Patients and caregivers should also be informed that only trained and certified prescribers, and healthcare facilities enrolled in the program are able to prescribe, dispense or administer LUMIZYME, and that patients must be enrolled in and meet all the conditions of the Lumizyme ACE Program to receive Lumizyme.
17.2 Pompe Registry
Patients and their caregivers should be informed that a registry for patients with Pompe disease (the Pompe Registry) has been established in order to better understand the variability and progression of Pompe disease, and to continue to monitor and evaluate long-term treatment effects of LUMIZYME. The Pompe Registry will also monitor the effect of LUMIZYME on pregnant women and their offspring [see Use in Specific Populations (8)]. Patients and their caregivers are encouraged to participate in the Pompe Registry and advised that their participation is voluntary and may involve long-term follow-up. For more information regarding the registry program visit www.pomperegistry.com or by calling 1-800-745-4447.
17.3 Infusion Reactions
Patients and caregivers should be informed that the most common adverse reactions observed with LUMIZYME were infusion reactions. Infusion reactions may occur during or within 2 hours after completion of the infusion. Symptoms associated with infusion reactions include urticaria, diarrhea, vomiting, dyspnea, pruritus, rash/erythema, pharyngolaryngeal pain, neck pain, hypoacusis, flushing/feeling hot, pain in extremity, fall and chest discomfort, respiratory distress, cough, livedo reticularis, agitation, irritability, retching, rigors, tremor and increased lacrimation.
LUMIZYME is manufactured and distributed by:
Genzyme Corporation
500 Kendall Street
Cambridge, MA 02142
1-800-745-4447 (phone)
US License Number: 1596
LUMIZYME is a trade mark of Genzyme Corporation.
Genzyme is a registered trademark of Genzyme Corporation.
Lumizyme Ace Program is a Service Mark of Genzyme Corporation.
Package Carton – Principal Display Panel – 50 mg Carton
Package contains one vial of
Lumizyme™
(alglucosidase alfa)
50 mg/vial
Store Refrigerated At 2-8°C (36-46°F)
Do Not Freeze or Shake
Protect From Light
Contains No Preservatives
For Single Use Only
No U.S. Standard of Potency
See package insert for
dosage and administration.
Genzyme

| LUMIZYME
alglucosidase alfa injection, powder, for solution |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA125291 | 05/24/2010 | |
| Labeler - Genzyme Corporation (025322157) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Genzyme Flanders BVBA | 372153895 | MANUFACTURE, ANALYSIS, API MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Genzyme Ireland Ltd | 985127419 | ANALYSIS, MANUFACTURE, PACK, LABEL | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Genzyme Corporation | 034378252 | LABEL, PACK, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Genzyme Limited | 229522842 | PACK, LABEL | |