FLUOROURACIL
-
fluorouracil injection, solution
Sandoz Inc.
----------
Fluorouracil Injection,USP
Rx Only
WARNING
It is recommended that Fluorouracil Injection be given only by or under the supervision of a qualified physician who is experienced in cancer chemotherapy and who is well versed in the use of potent antimetabolites. Because of the possibility of severe toxic reactions, it is recommended that patients be hospitalized at least during the initial course of therapy.
These instructions should be thoroughly reviewed before administration of Fluorouracil Injection, USP.
DESCRIPTION
Fluorouracil Injection, USP an antineoplastic antimetabolite, is a sterile, nonpyrogenic injectable solution for intravenous administration. Each mL contains 50 mg of fluorouracil and water for injection. Sodium hydroxide may be added to adjust pH to approximately 9.2 during manufacture.
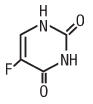
Chemically, fluorouracil, a fluorinated pyrimidine, is 5-fluoro-2,4 (1H,3H)-pyrimidinedione. It is a white to practically white crystalline powder which is sparingly soluble in water. The molecular weight of fluorouracil is 130.08.
Molecular formula of fluorouracil is: C4H3FN2O2.
The structural formula is:
CLINICAL PHARMACOLOGY
There is evidence that the metabolism of fluorouracil in the anabolic pathway blocks the methylation reaction of deoxyuridylic acid to thymidylic acid. In this manner, fluorouracil interferes with the synthesis of deoxyribonucleic acid (DNA) and to a lesser extent inhibits the formation of ribonucleic acid (RNA). Since DNA and RNA are essential for cell division and growth, the effect of fluorouracil may be to create a thymine deficiency which provokes unbalanced growth and death of the cell. The effects of DNA and RNA deprivation are most marked on those cells which grow more rapidly and which take up fluorouracil at a more rapid rate.
Following intravenous injection,fluorouracil distributes into tumors, intestinal mucosa, bone marrow, liver and other tissues throughout the body. In spite of its limited lipid solubility, fluorouracil diffuses readily across the blood-brain barrier and distributes into cerebrospinal fluid and brain tissue.
Seven percent to 20% of the parent drug is excreted unchanged in the urine in 6 hours; of this over 90% is excreted in the first hour. The remaining percentage of the administered dose is metabolized, primarily in the liver. The catabolic metabolism of fluorouracil results in degradation products (e.g., CO2, urea and α-fluoro-β-alanine) which are inactive. The inactive metabolites are excreted in the urine over the next 3 to 4 hours. When fluorouracil is labeled in the six carbon position, thus preventing the 14C metabolism to CO2, approximately 90% of the total radioactivity is excreted in the urine. When fluorouracil is labeled in the two carbon position approximately 90% of the total radioactivity is excreted in expired CO2. Ninety percent of the dose is accounted for during the first 24 hours following intravenous administration.
Following intravenous administration of fluorouracil, the mean half-life of elimination from plasma is approximately 16 minutes, with a range of 8 to 20 minutes, and is dose dependent. No intact drug can be detected in the plasma 3 hours after an intravenous injection.
INDICATIONS AND USAGE
Fluorouracil Injection, USP is effective in the palliative management of carcinoma of the colon, rectum, breast, stomach and pancreas.
CONTRAINDICATIONS
Fluorouracil therapy is contraindicated for patients in a poor nutritional state, those with depressed bone marrow function, those with potentially serious infections or those with a known hypersensitivity to Fluorouracil.
WARNINGS
(see BOXED WARNING)
THE DAILY DOSE OF FLUOROURACIL INJECTION IS NOT TO EXCEED 800 MG. IT IS RECOMMENDED THAT PATIENTS BE HOSPITALIZED DURING THEIR FIRST COURSE OF TREATMENT.
Fluorouracil should be used with extreme caution in poor risk patients with a history of high-dose pelvic irradiation or previous use of alkylating agents, those who have a widespread involvement of bone marrow by metastatic tumors or those with impaired hepatic or renal function.
Rarely, unexpected, severe toxicity (e.g., stomatitis, diarrhea, neutropenia and neurotoxicity) associated with 5-fluorouracil has been attributed to deficiency of dipyrimidine dehydrogenase activity.1 A few patients have been rechallenged with 5-fluorouracil and despite 5-fluorouracil dose lowering, toxicity recurred and progressed with worse morbidity. Absence of this catabolic enzyme appears to result in prolonged clearance of 5-fluorouracil.
Pregnancy
Teratogenic Effects – Pregnancy Category D
Fluorouracil may cause fetal harm when administered to a pregnant woman. Fluorouracil has been shown to be teratogenic in laboratory animals. Fluorouracil exhibited maximum teratogenicity when given to mice as single intraperitoneal injections of 10 to 40 mg/kg on day 10 or 12 of gestation. Similarly, intraperitoneal doses of 12 to 37 mg/kg given to rats between days 9 and 12 of gestation and intramuscular doses of 3 to 9 mg given to hamsters between days 8 and 11 of gestation were teratogenic. Malformations included cleft palates, skeletal defects and deformed appendages, paws and tails. The dosages which were teratogenic in animals are 1 to 3 times the maximum recommended human therapeutic dose. In monkeys, divided doses of 40 mg/kg given between days 20 and 24 of gestation were not teratogenic.
There are no adequate and well-controlled studies with fluorouracil in pregnant women. While there is no evidence of teratogenicity in humans due to fluorouracil, it should be kept in mind that other drugs which inhibit DNA synthesis (e.g., methotrexate and aminopterin) have been reported to be teratogenic in humans. Women of childbearing potential should be advised to avoid becoming pregnant. If the drug is used during pregnancy, or if the patient becomes pregnant while taking the drug, the patient should be told of the potential hazard to the fetus. Fluorouracil should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Combination Therapy
Any form of therapy which adds to the stress of the patient, interferes with nutrition or depresses bone marrow function will increase the toxicity of fluorouracil.
PRECAUTIONS
General
Fluorouracil is a highly toxic drug with a narrow margin of safety. Therefore, patients should be carefully supervised, since therapeutic response is unlikely to occur without some evidence of toxicity. Severe hematological toxicity, gastrointestinal hemorrhage and even death may result from the use of fluorouracil despite meticulous selection of patients and careful adjustment of dosage. Although severe toxicity is more likely in poor risk patients, fatalities may be encountered occasionally even in patients in relatively good condition.
Therapy is to be discontinued promptly whenever one of the following signs of toxicity appears:
- Stomatitis or esophagopharyngitis, at the first visible sign.
- Leukopenia (WBC under 3500) or a rapidly falling white blood count.
- Vomiting, intractable.
- Diarrhea, frequent bowel movements or watery stools.
- Gastrointestinal ulceration and bleeding.
- Thrombocytopenia (platelets under 100,000).
- Hemorrhage from any site.
The administration of 5-fluorouracil has been associated with the occurrence of palmar-plantar erythrodysesthesia syndrome, also known as hand-foot syndrome. This syndrome has been characterized as a tingling sensation of hands and feet which progress over the next few days to pain when holding objects or walking. The palms and soles became symmetrically swollen and erythematous with tenderness of the distal phalanges, possibly accompanied by desquamation. Interruption of therapy is followed by gradual resolution over 5 to 7 days. Although pyridoxine has been reported to ameliorate the palmarplantar erythrodysesthesia syndrome, its safety and effectiveness has not been established.
Information for Patients
Patients should be informed of expected toxic effects, particularly oral manifestations. Patients should be alerted to the possibility of alopecia as a result of therapy and should be informed that it is usually a transient effect.
Laboratory Tests
White blood counts with differential are recommended before each dose
Drug Interactions
Leucovorin calcium may enhance the toxicity of fluorouracil.
Also see WARNINGS section.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term studies in animals to evaluate the carcinogenic potential of fluorouracil have not been conducted. However, there was no evidence of carcinogenicity in small groups of rats given fluorouracil orally at doses of 0.01, 0.3, 1 or 3 mg per rat 5 days per week for 52 weeks, followed by a six-month observation period. Also, in other studies, 33 mg/kg of fluorouracil was administered intravenously to male rats once a week for 52 weeks followed by observation for the remainder of their lifetimes with no evidence of carcinogenicity. Female mice were given 1 mg of fluorouracil intravenously once a week for 16 weeks with no effect on the incidence of lung adenomas. On the basis of the available data, no evaluation can be made of the carcinogenic risk of fluorouracil to humans.
Mutagenesis
Oncogenic transformation of fibroblasts from mouse embryo has been induced in vitro by fluorouracil, but the relationship between oncogenicity and mutagenicity is not clear. Fluorouracil has been shown to be mutagenic to several strains of Salmonella typhimurium, including TA 1535, TA 1537 and TA 1538, and to Saccharomyces cerevisiae, although no evidence of mutagenicity was found with Salmonella typhimurium strains TA 92, TA 98 and TA 100. In addition, a positive effect was observed in the micronucleus test on bone marrow cells of the mouse, and fluorouracil at very high concentrations produced chromosomal breaks in hamster fibroblasts in vitro.
Impairment of Fertility
Fluorouracil has not been adequately studied in animals to permit an evaluation of its effects on fertility and general reproductive performance. However, doses of 125 or 250 mg/kg, administered intraperitoneally, have been shown to induce chromosomal aberrations and changes in chromosomal organization of spermatogonia in rats. Spermatogonial differentiation was also inhibited by fluorouracil, resulting in transient infertility. However, in studies with a strain of mouse which is sensitive to the induction of sperm head abnormalities after exposure to a range of chemical mutagens and carcinogens, fluorouracil did not produce any abnormalities at oral doses of up to 80 mg/kg/day. In female rats, fluorouracil, administered intraperitoneally at weekly doses of 25 or 50 mg/kg for 3 weeks during the pre-ovulatory phases of oogenesis, significantly reduced the incidence of fertile matings, delayed the development of pre- and post-implantation embryos, increased the incidence of pre-implantation lethality and induced chromosomal anomalies in these embryos. In a limited study in rabbits, a single 25 mg/kg dose of fluorouracil or 5 daily doses of 5 mg/kg had no effect on ovulation, appeared not to affect implantation and had only a limited effect in producing zygote destruction. Compounds such as fluorouracil, which interfere with DNA, RNA and protein synthesis, might be expected to have adverse effects on gametogenesis.
Pregnancy
Pregnancy Category D. See WARNINGS section.
Nonteratogenic Effects
Fluorouracil has not been studied in animals for its effects on peri- and postnatal development. However, fluorouracil has been shown to cross the placenta and enter into fetal circulation in the rat. Administration of fluorouracil has resulted in increased resorptions and embryolethality in rats. In monkeys, maternal doses higher than 40 mg/kg resulted in abortion of all embryos exposed to fluorouracil. Compounds which inhibit DNA, RNA and protein synthesis might be expected to have adverse effects on peri- and postnatal development.
Nursing Mothers
It is not known whether fluorouracil is excreted in human milk. Because fluorouracil inhibits DNA, RNA and protein synthesis, mothers should not nurse while receiving this drug.
Pediatric Use
Safety and effectiveness in children have not been established.
ADVERSE REACTIONS
Stomatitis and esophagopharyngitis (which may lead to sloughing and ulceration), diarrhea, anorexia, nausea and emesis are commonly seen during therapy.
Leukopenia usually follows every course of adequate therapy with fluorouracil. The lowest white blood cell counts are commonly observed between the 9th and 14th days after the first course of treatment, although uncommonly the maximal depression may be delayed for as long as 20 days. By the 30th day the count has usually returned to the normal range.
Alopecia and dermatitis may be seen in a substantial number of cases. The dermatitis most often seen is a pruritic maculopapular rash usually appearing on the extremities and less frequently on the trunk. It is generally reversible and usually responsive to symptomatic treatment.
Other adverse reactions are:
Hematologic: pancytopenia, thrombocytopenia, agranulocytosis, anemia.
Cardiovascular: myocardial ischemia, angina.
Gastrointestinal: gastrointestinal ulceration and bleeding.
Allergic Reactions: anaphylaxis and generalized allergic reactions.
Neurologic: acute cerebellar syndrome (which may persist following discontinuance of treatment), nystagmus, headache.
Dermatologic: dry skin; fissuring; photosensitivity, as manifested by erythema or increased pigmentation of the skin; vein pigmentation; palmar-plantar erythrodysesthesia syndrome, as manifested by tingling of the hands and feet followed by pain, erythema and swelling.
Ophthalmic: lacrimal duct stenosis, visual changes, lacrimation, photophobia.
Psychiatric: disorientation, confusion, euphoria.
Miscellaneous: thrombophlebitis, epistaxis, nail changes (including loss of nails).
OVERDOSAGE
The possibility of overdosage with fluorouracil is unlikely in view of the mode of administration. Nevertheless, the anticipated manifestations would be nausea, vomiting, diarrhea, gastrointestinal ulceration and bleeding, bone marrow depression (including thrombocytopenia, leukopenia and agranulocytosis). No specific antidotal therapy exists. Patients who have been exposed to an overdose of fluorouracil should be monitored hematologically for at least four weeks. Should abnormalities appear, appropriate therapy should be utilized.
The acute intravenous toxicity of fluorouracil is as follows:
| Species | LD50
(mg/kg ±S.E.) |
|---|---|
| Mouse | 340 ± 17 |
| Rat | 165 ± 26 |
| Rabbit | 27 ± 5.1 |
| Dog | 31.5 ± 3.8 |
DOSAGE AND ADMINISTRATION
General Instructions
Fluorouracil Injection, USP should be administered only intravenously, using care to avoid extravasation. No dilution is required.
All dosages are based on the patient's actual weight. However, the estimated lean body mass (dry weight) is used if the patient is obese or if there has been a spurious weight gain due to edema, ascites or other forms of abnormal fluid retention.
It is recommended that prior to treatment each patient be carefully evaluated in order to estimate as accurately as possible the optimum initial dosage of Fluorouracil.
Dosage
12 mg/kg are given intravenously once daily for 4 successive days. The daily dose should not exceed 800 mg. If no toxicity is observed, 6 mg/kg are given on the 6th, 8th, 10th and 12th days unless toxicity occurs. No therapy is given on the 5th, 7th, 9th or 11th days. Therapy is to be discontinued at the end of the 12th day, even if no toxicity has become apparent. (See WARNINGS and PRECAUTIONS sections.)
Poor risk patients or those who are not in an adequate nutritional state (see CONTRAINDICATIONS and WARNINGS sections) should receive 6 mg/kg/day for 3 days. If no toxicity is observed, 3 mg/kg may be given on the 5th, 7th and 9th days unless toxicity occurs. No therapy is given on the 4th, 6th or 8th days. The daily dose should not exceed 400 mg.
A sequence of injections on either schedule constitutes a "course of therapy."
Maintenance Therapy
In instances where toxicity has not been a problem, it is recommended that therapy be continued using either of the following schedules:
- Repeat dosage of first course every 30 days after the last day of the previous course of treatment.
- When toxic signs resulting from the initial course of therapy have subsided, administer a maintenance dosage of 10 to 15 mg/kg/week as a single dose. Do not exceed 1 gm per week.
The patient's reaction to the previous course of therapy should be taken into account in determining the amount of the drug to be used, and the dosage should be adjusted accordingly. Some patients have received from 9 to 45 courses of treatment during periods which ranged from 12 to 60 months.
Handling and Disposal
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published.2–7 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
Note: Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Although the Fluorouracil solution may discolor slightly during storage, the potency and safety are not adversely affected. If a precipitate occurs due to exposure to low temperatures, resolubilize by heating to 140°F and shaking vigorously; allow to cool to body temperature before using.
HOW SUPPLIED
Fluorouracil Injection, USP is available for intravenouse use in 10 mL vials. Each mL contains 50 mg (available as 500 mg/10 mL) of fluorouracil in a colorless to faint yellow aqueous solution. Sodium hydroxide may have been added to adjust the pH to approximately 9.2 during manufacture.
NDC 66758-044-03: Carton of 10 × 10 mL single use vials
STORAGE
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature]. Do not refrigerate or freeze. Protect from light. Retain in carton until contents are used. Discard any unused portion.
For Parenta Customer Service, call 1-800-898-9948.
Manufactured for:
PARENTA®
pharmaceuticals
an Ebewe Pharma Company
Yardley, PA 19067
Manufactured by:
Ebewe
PHARMA
A-4866 Unterach,
AUSTRIA
April 2009
782592–03
REFERENCES
- Harris BE, Carpenter JT, Diasio RB: Severe 5-Fluorouracil Toxicity Secondary to Dihydropyrimidine Dehydrogenase Deficiency. A potentially more common phramacologic syndrome. Cancer. August 1, 1991; 68:499–501.
- Recommendations for the Safe Handling of Parenteral Antineoplastic Drugs. Washington, DC, U.S. Government Printing Office (NIH Publication No. 83-2621).
- AMA Council Report. Guidelines for Handling Parenteral Antineoplastics. JAMA Mar 15, 1985; 253:1590–1592.
- National Study Commission on Cytotoxic Exposure: Recommendations for Handling Cytotoxic Agents. Available from Louis P. Jeffrey, ScD, Director of Pharmacy Services, Rhode Island Hospital, 593 Eddy Street, Providence, Rhode Island 02902.
- Clinical Oncological Society of Australia: Guidelines and Recommendations for Safe Handling of Antineoplastic Agents. Med J Aust. Apr 30, 1983; 1:426–428.
- Jones RB, Frank R, Mass T: Safe Handling of Chemotherapeutic Agents: A Report from the Mount Sinai Medical Center. CA. Sept–Oct 1983; 33:258–263.
- American Society of Hospital Pharmacists Technical Assistance Bulletin on Handling Cytotoxic Drugs in Hospitals. Am J Hosp Pharm. Jan 1985; 42:131–137.
- OSHA Work-Practice Guidelines for Personnel Dealing with Cytotoxic (Antineoplastic) Drugs. Am J Hosp Pharm. 1986; 43:1193–1204.
PRINCIPAL DISPLAY PANEL- 10 mL Vial Carton
NDC 66758-044-03
10 x 10 mL Single Use Vials
FLUOROURACIL INJECTION USP
500 mg/10 mL
(50 mg/mL)
For Intravenous Use Only.
Caution: Cytotoxic Agent
Rx only
PARENTA®
pharmaceuticals
an Ebewe Pharma Company

| FLUOROURACIL
fluorouracil injection, solution |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| ANDA | ANDA040772 | 09/10/2008 | |
| Labeler - Sandoz Inc. (005387188) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Ebewe Pharma Ges.m.b.H. Nfg.KG | 300529422 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Labor L+S AG | 313710642 | Analysis | |