GAMUNEX - human immunoglobulin g injection
GRIFOLS USA, LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use GAMUNEX®, Immune Globulin Intravenous (Human), 10% Caprylate/Chromatography Purified, safely and effectively. See full prescribing information for GAMUNEX.
GAMUNEX (Immune Globulin Intravenous [Human], 10% Caprylate/Chromatography Purified) 10% Liquid Preparation Initial U.S. Approval 2003 WARNING: ACUTE RENAL DYSFUNCTION and FAILURESee full prescribing information for complete boxed warning.
RECENT MAJOR CHANGESINDICATIONS AND USAGEDOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSGAMUNEX is supplied in 1 g, 2.5 g, 5 g, 10 g, or 20 g single use bottles. (3)
CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact: Talecris Biotherapeutics at 1-800-520-2807, or FDA at 1-800-FDA-1088 or go to www.fda.gov/medwatch DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 10/2008 |
FULL PRESCRIBING INFORMATION
WARNING: ACUTE RENAL DYSFUNCTION AND ACUTE RENAL FAILURE
Immune Globulin Intravenous (Human) products have been reported to be associated with renal dysfunction, acute renal failure, osmotic nephrosis and death.(1) Patients predisposed to acute renal failure include patients with any degree of pre-existing renal insufficiency, diabetes mellitus, age greater than 65, volume depletion, sepsis, paraproteinemia, or patients receiving known nephrotoxic drugs. Especially in such patients, IGIV products should be administered at the minimum concentration available and the minimum rate of infusion practicable. While these reports of renal dysfunction and acute renal failure have been associated with the use of many of the licensed IGIV products, those containing sucrose as a stabilizer accounted for a disproportionate share of the total number. GAMUNEX does not contain sucrose. Glycine, a natural amino acid, is used as a stabilizer. (See Dosage and Administration [2.5] and Warnings and Precautions [5.2] for important information intended to reduce the risk of acute renal failure.)
1 INDICATIONS AND USAGE
Gamunex is an immune globulin intravenous (human) 10% liquid indicated for the treatment of:
1.1 Primary Humoral Immunodeficiency (PI)
GAMUNEX is indicated as replacement therapy of primary humoral immunodeficiency. This includes, but is not limited to, congenital agammaglobulinemia, common variable immunodeficiency, X-linked agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.(2-9)
1.2 Idiopathic Thrombocytopenic Purpura (ITP)
GAMUNEX is indicated in Idiopathic Thrombocytopenic Purpura to rapidly raise platelet counts to prevent bleeding or to allow a patient with ITP to undergo surgery.(10-15)
2 DOSAGE AND ADMINISTRATION
For Intravenous use only.
GAMUNEX consists of 9%-11% protein in 0.16-0.24 M glycine. The buffering capacity of GAMUNEX is 35.0 mEq/L (0.35 mEq/g protein). A dose of 1 g/kg body weight therefore represents an acid load of 0.35 mEq/kg body weight. The total buffering capacity of whole blood in a normal individual is 45-50 mEq/L of blood, or 3.6 mEq/kg body weight.(16) Thus, the acid load delivered with a dose of 1 g/kg of GAMUNEX would be neutralized by the buffering capacity of whole blood alone, even if the dose was infused instantaneously.
2.1 Preparation and Handling
- GAMUNEX should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if turbid.
- Do not freeze. Solutions that have been frozen should not be used.
- The GAMUNEX vial is for single use only. GAMUNEX contains no preservative. Any vial that has been entered should be used promptly. Partially used vials should be discarded.
- GAMUNEX should be infused using a separate line by itself, without mixing with other intravenous fluids or medications the subject might be receiving.
- GAMUNEX is not compatible with saline. If dilution is required, GAMUNEX may be diluted with 5% dextrose in water (D5/W). No other drug interactions or compatibilities have been evaluated.
- Content of vials may be pooled under aseptic conditions into sterile infusion bags and infused within 8 hours after pooling.
- Do not mix with immune globulin intravenous (IGIV) products from other manufacturers.
- Do not use after expiration date.
2.2 Treatment of Primary Humoral Immunodeficiency
As there are significant differences in the half-life of IgG among patients with primary immunodeficiencies, the frequency and amount of immunoglobulin therapy may vary from patient to patient. The proper amount can be determined by monitoring clinical response.
The dose of GAMUNEX for replacement therapy in primary immune deficiency diseases is 300 to 600 mg/kg body weight (3-6 mL/kg) administered every 3 to 4 weeks. The dosage may be adjusted over time to achieve the desired trough levels and clinical responses.
If a patient routinely receives a dose of less than 400 mg/kg of GAMUNEX every 3 to 4 weeks (less than 4 mL/kg), and is at risk of measles exposure (i.e., traveling to a measles endemic area), administer a dose of at least 400 mg/kg (4 mL/kg) just prior to the expected measles exposure. If a patient has been exposed to measles, a dose of 400 mg/kg (4 mL/kg) should be administered as soon as possible after exposure.
2.3 Treatment of Idiopathic Thrombocytopenic Purpura
GAMUNEX may be administered at a total dose of 2 g/kg, divided in two doses of 1 g/kg (10 mL/kg) given on two consecutive days or into five doses of 0.4 g/kg (4 mL/kg) given on five consecutive days. If after administration of the first of two daily 1 g/kg (10 mL/kg) doses, an adequate increase in the platelet count is observed at 24 hours, the second dose of 1g/kg body weight may be withheld.
Forty-eight ITP subjects were treated with 2 g/kg GAMUNEX, divided in two 1 g/kg doses (10 mL/kg) given on two successive days. With this dose regimen 35/39 subjects (90%) responded with a platelet count from less than or equal to 20 ×109/L to more than or equal to 50 ×109/L within 7 days after treatment.(17)
The high dose regimen (1 g/kg × 1-2 days) is not recommended for individuals with expanded fluid volumes or where fluid volume may be a concern.
2.4 Treatment of Chronic Inflammatory Demyelinating Polyneuropathy
GAMUNEX may be initially administered as a total loading dose of 2 g/kg (20 mL/kg) given in divided doses over two to four consecutive days. GAMUNEX may be administered as a maintenance infusion of 1 g/kg (10 mL/kg) administered over 1 day or divided into two doses of 0.5 g/kg (5 mL/kg) given on two consecutive days, every 3 weeks.
2.5 Administration
GAMUNEX should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if turbid and/or if discoloration is observed.
Only administer intravenously. GAMUNEX should be at room temperature during administration.
Only 18 gauge needles should be used to penetrate the stopper for dispensing product from the 10 mL vial; 16 gauge needles or dispensing pins should only be used with 25 mL vial sizes and larger. Needles or dispensing pins should only be inserted once and be within the stopper area delineated by the raised ring. The stopper should be penetrated perpendicular to the plane of the stopper within the ring.
| GAMUNEX® vial size | Gauge of needle to penetrate stopper |
| 10 mL | 18 gauge |
| 25, 50, 100, 200 mL | 16 gauge |
Any vial that has been opened should be used promptly. Partially used vials should be discarded.
If dilution is required, GAMUNEX may be diluted with 5% dextrose in water (D5/W).
Rate of Administration
It is recommended that GAMUNEX should initially be infused at a rate of 0.01 mL/kg per minute (1 mg/kg per minute) for the first 30 minutes.
If well-tolerated, the rate may be gradually increased to a maximum of 0.08 mL/kg per minute (8 mg/kg per minute).
| Indication | Initial infusion rate (first 30 minutes) | Maximum infusion rate (if tolerated) |
| PI | 1 mg/kg/min | 8 mg/kg/min |
| ITP | 1 mg/kg/min | 8 mg/kg/min |
| CIDP | 2 mg/kg/min | 8 mg/kg/min |
Certain severe adverse drug reactions may be related to the rate of infusion. Slowing or stopping the infusion usually allows the symptoms to disappear promptly.
Ensure that patients with pre-existing renal insufficiency are not volume depleted; discontinue GAMUNEX if renal function deteriorates.
For patients at risk of renal dysfunction or thromboembolic events, administer GAMUNEX at the minimum infusion rate practicable.
Incompatibilities
GAMUNEX is not compatible with saline. If dilution is required, GAMUNEX may be diluted with 5% dextrose in water (D5/W). No other drug interactions or compatibilities have been evaluated.
3 DOSAGE FORMS AND STRENGTH
GAMUNEX is supplied in 1 g, 2.5 g, 5 g, 10 g, or 20 g single use bottles.
- 1 g in 10 mL solution
- 2.5 g in 25 mL solution
- 5 g in 50 mL solution
- 10 g in 100 mL solution
- 20 g in 200 mL solution
4 CONTRAINDICATIONS
- GAMUNEX is contraindicated in individuals with acute severe hypersensitivity reactions to Immune Globulin (Human).
- GAMUNEX contains trace amounts of IgA. It is contraindicated in IgA deficient patients with antibodies against IgA and history of hypersensitivity. (See Description [11] )
5 WARNINGS AND PRECAUTIONS
5.1 Sensitivity
Severe hypersensitivity reactions may occur. In case of hypersensitivity, IGIV infusion should be immediately discontinued and appropriate treatment instituted. Epinephrine should be immediately available for treatment of acute severe hypersensitivity reaction. (See Patient Counseling Information [17])
GAMUNEX contains trace amounts of IgA (average 46 micrograms/mL). It is contraindicated in IgA deficient patients with antibodies against IgA and history of hypersensitivity. (See Patient Counseling Information [17])
5.2 Renal Failure
Assure that patients are not volume depleted prior to the initiation of the infusion of IGIV. Periodic monitoring of renal function and urine output is particularly important in patients judged to have a potential increased risk for developing acute renal failure. Renal function, including measurement of blood urea nitrogen (BUN)/serum creatinine, should be assessed prior to the initial infusion of GAMUNEX and again at appropriate intervals thereafter. If renal function deteriorates, discontinuation of the product should be considered. (See Patient Counseling Information [17]) For patients judged to be at risk for developing renal dysfunction and/or at risk of developing thrombotic events, it may be prudent to reduce the amount of product infused per unit time by infusing GAMUNEX at a rate less than 8 mg IG/kg/min (0.08 mL/kg/min). (See Boxed Warning) (See Dosage and Administration [2.5])
5.3 Hyperproteinemia
Hyperproteinemia, increased serum viscosity and hyponatremia may occur in patients receiving IGIV therapy. The hyponatremia is likely to be a pseudohyponatremia as demonstrated by a decreased calculated serum osmolality or elevated osmolar gap. Distinguishing true hyponatremia from pseudohyponatremia is clinically critical, as treatment aimed at decreasing serum free water in patients with pseudohyponatremia may lead to volume depletion, a further increase in serum viscosity and a disposition to thromboembolic events.(18)
5.4 Thrombotic Events
Thrombotic events have been reported in association with IGIV.(19-21) Patients at risk may include those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, coagulation disorders, prolonged periods of immobilization and/or known or suspected hyperviscosity. The potential risks and benefits of IGIV should be weighed against those of alternative therapies for all patients for whom IGIV administration is being considered. Baseline assessment of blood viscosity should be considered in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies.
5.5 Aseptic Meningitis Syndrome (AMS)
An aseptic meningitis syndrome (AMS) has been reported to occur infrequently in association with Immune Globulin Intravenous (Human) treatment. Discontinuation of IGIV treatment has resulted in remission of AMS within several days without sequelae.(22-24) The syndrome usually begins within several hours to two days following IGIV treatment. It is characterized by symptoms and signs including severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea and vomiting. Cerebrospinal fluid (CSF) studies are frequently positive with pleocytosis up to several thousand cells per cu mm, predominantly from the granulocytic series, and elevated protein levels up to several hundred mg/dl. Patients exhibiting such symptoms and signs should receive a thorough neurological examination, including CSF studies, to rule out other causes of meningitis. It appears that patients with a history of migraine may be more susceptible. (See Patient Counseling Information [17])
5.6 Hemolysis
Immune Globulin Intravenous (Human) (IGIV) products can contain blood group antibodies which may act as hemolysins and induce in vivo coating of red blood cells with immunoglobulin, causing a positive direct antiglobulin reaction and, rarely, hemolysis.(25–27) Hemolytic anemia can develop subsequent to IGIV therapy due to enhanced RBC sequestration. IGIV recipients should be monitored for clinical signs and symptoms of hemolysis.(28) If signs and/or symptoms of hemolysis are present after IGIV infusion, appropriate confirmatory laboratory testing should be done. (See Patient Counseling Information [17])
5.7 Transfusion-related Acute Lung Injury (TRALI)
There have been reports of noncardiogenic pulmonary edema [Transfusion-Related Acute Lung Injury (TRALI)] in patients administered IGIV.(29) TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever and typically occurs within 1-6 hrs after transfusion. Patients with TRALI may be managed using oxygen therapy with adequate ventilatory support.
IGIV recipients should be monitored for pulmonary adverse reactions. (See Patient Counseling Information [17]) If TRALI is suspected, appropriate tests should be performed for the presence of anti-neutrophil antibodies in both the product and patient serum.
5.8 Volume Overload
The high dose regimen (1 g/kg × 1-2 days) is not recommended for individuals with expanded fluid volumes or where fluid volume may be a concern.
5.9 General
Because this product is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, and, theoretically, the Creutzfeldt-Jakob (CJD) agent. ALL infections thought by a physician possibly to have been transmitted by this product should be reported by the physician or other healthcare provider to Talecris Biotherapeutics, Inc. [1-800-520-2807]. The physician should discuss the risks and benefits of this product with the patient, before prescribing or administering it to the patient. (See Patient Counseling Information [17])
5.10 Laboratory Tests
If signs and/or symptoms of hemolysis are present after IGIV infusion, appropriate confirmatory laboratory testing should be done.
If TRALI is suspected, appropriate tests should be performed for the presence of anti-neutrophil antibodies in both the product and patient serum.
Because of the potentially increased risk of thrombosis, baseline assessment of blood viscosity should be considered in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies.
6 ADVERSE REACTIONS
6.1 Adverse Drug Reaction Overview
The most serious adverse reaction observed in clinical study subjects receiving GAMUNEX for PI was an exacerbation of autoimmune pure red cell aplasia in one subject.
The most serious adverse reaction observed in clinical study subjects receiving GAMUNEX for ITP was myocarditis in one subject that occurred 50 days post study drug infusion and was not considered drug related.
The most serious adverse reaction observed in clinical study subjects receiving GAMUNEX for CIDP was pulmonary embolism (PE) in one subject with a history of PE.
The most common drug related adverse reactions observed at a rate ≥5% in subjects with PI were headache, cough, injection site reaction, nausea, pharyngitis and urticaria.
The most common drug related adverse reactions observed at a rate ≥5% in subjects with ITP were headache, vomiting, fever, nausea, back pain and rash.
The most common drug related adverse reactions observed at a rate ≥5% in subjects with CIDP were headache, fever, chills, hypertension, rash, nausea and asthenia.
6.2 Clinical Trials Adverse Drug Reactions
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in practice.
Adverse events similar to those previously reported with the administration of intravenous and intramuscular immunoglobulin products may occur. Cases of reversible aseptic meningitis, migraine, isolated cases of reversible hemolytic anemia and reversible increases in liver function tests have been observed with GAMUNEX. Immediate anaphylactic reactions can possibly occur (<0.01%). Epinephrine should be available for treatment of any acute anaphylactoid reaction. (see Warnings and Precautions [5.1])
Treatment of Primary Humoral Immunodeficiency
The following table shows the number of subjects treated with GAMUNEX in clinical trials to study PI, and the reason for discontinuation due to adverse events:
|
|||
| Study Number | Number of Subjects Treated with GAMUNEX® | Number of Subjects Discontinued Due to Adverse Events | Adverse Event |
| 100152 | 18 | 0 | ----- |
| 100174 | 20 | 1 | Coombs negative hypochromic anemia* |
| 100175 | 87 | 1 | Autoimmune pure red cell aplasia* |
In study 100175, 9 subjects in each treatment group were pretreated with non-steroidal medication prior to infusion. Generally, diphenhydramine and acetaminophen were used.
Any adverse events in trial 100175, irrespective of the causality assessment, are given in the following table.
| Adverse Event | GAMUNEX®
No. of subjects: 87 No. of subjects with AE (percentage of all subjects) | GAMIMUNE® N, 10% No. of subjects: 85 No. of subjects with AE (percentage of all subjects) |
| Cough increased | 47 (54%) | 46 (54%) |
| Rhinitis | 44 (51%) | 45 (53%) |
| Pharyngitis | 36 (41%) | 39 (46%) |
| Headache | 22 (25%) | 28 (33%) |
| Fever | 24 (28%) | 27 (32%) |
| Diarrhea | 24 (28%) | 27 (32%) |
| Asthma | 25 (29%) | 17 (20%) |
| Nausea | 17 (20%) | 22 (26%) |
| Ear Pain | 16 (18%) | 12 (14%) |
| Asthenia | 9 (10%) | 13 (15%) |
The subset of drug related adverse events in trial 100175 reported by at least 5% of subjects during the 9-month treatment are given in the following table.
| Drug Related Adverse Event | GAMUNEX®
No. of subjects: 87 No. of subjects with drug related AE (percentage of all subjects) | GAMIMUNE® N, 10% No. of subjects: 85 No. of subjects with drug related AE (percentage of all subjects) |
| Headache | 7 (8%) | 8 (9%) |
| Cough increased | 6 (7%) | 4 (5%) |
| Injection site reaction | 4 (5%) | 7 (8%) |
| Nausea | 4 (5%) | 4 (5%) |
| Pharyngitis | 4 (5%) | 3 (4%) |
| Urticaria | 4 (5%) | 1 (1%) |
Adverse events, which were reported by at least 5% of subjects were also analyzed by frequency and in relation to infusions administered. The analysis is displayed in the following table.
| Adverse Event | GAMUNEX® | GAMIMUNE® N, 10% | |
| No. of infusions: 825 | No. of infusions: 865 | ||
| No. of AE | No. of AE | ||
| (percentage of all infusions) | (percentage of all infusions) | ||
| Cough increased | All | 154 (18.7%) | 148 (17.1%) |
| Drug related | 14 (1.7%) | 11 (1.3%) | |
| Pharyngitis | All | 96 (11.6%) | 99 (11.4%) |
| Drug related | 7 (0.8%) | 9 (1.0%) | |
| Headache | All | 57 (6.9%) | 69 (8.0%) |
| Drug related | 7 (0.8%) | 11 (1.3%) | |
| Fever | All | 41 (5.0%) | 65 (7.5%) |
| Drug related | 1 (0.1%) | 9 (1.0%) | |
| Nausea | All | 31 (3.8%) | 43 (5.0%) |
| Drug related | 4 (0.5%) | 4 (0.5%) | |
| Urticaria | All | 5 (0.6%) | 8 (0.9%) |
| Drug related | 4 (0.5%) | 5 (0.6%) |
The mean number of adverse events per infusion that occurred during or on the same day as an infusion was 0.21 in both the GAMUNEX and GAMIMUNE® N, Immune Globulin Intravenous (Human), 10%, treatment groups.
In all three trials in primary humoral immundeficiencies, the maximum infusion rate was 0.08 mL/kg/min (8 mg/kg/min). The infusion rate was reduced for 11 of 222 exposed subjects (7 GAMUNEX, 4 GAMIMUNE N, 10%) at 17 occasions. In most instances, mild to moderate hives/urticaria, itching, pain or reaction at infusion site, anxiety or headache was the main reason. There was one case of severe chills. There were no anaphylactic or anaphylactoid reactions to GAMUNEX or GAMIMUNE N, 10%.
In trial 100175, serum samples were drawn to monitor the viral safety at baseline and one week after the first infusion (for parvovirus B19), eight weeks after first and fifth infusion, and 16 weeks after the first and fifth infusion of IGIV (for hepatitis C) and at any time of premature discontinuation of the study. Viral markers of hepatitis C, hepatitis B, HIV-1, and parvovirus B19 were monitored by nucleic acid testing (NAT, Polymerase Chain Reaction (PCR)), and serological testing. There were no treatment emergent findings of viral transmission for either GAMUNEX or GAMIMUNE N, 10%.(30–32)
Treatment of Idiopathic Thrombocytopenic Purpura
The following table shows the number of subjects treated with GAMUNEX in clinical trials to study ITP, and the reason for discontinuation due to adverse events:
| Study Number | Number of Subjects Treated with GAMUNEX® | Number of Subjects Discontinued Due to Adverse Events | Adverse Event |
| 100213 | 28 | 1 | Hives |
| 100176 | 48 | 1 | Headache, Fever, Vomiting |
One subject, a 10-year-old boy, died suddenly from myocarditis 50 days after his second infusion of GAMUNEX. The death was judged to be unrelated to GAMUNEX.
No pre-medication with corticosteroids was permitted by the protocol. Twelve (12) ITP subjects treated in each treatment group were pretreated with medication prior to infusion. Generally, diphenhydramine and/or acetaminophen were used. More than 90% of the observed drug related adverse events were of mild to moderate severity and of transient nature.
The infusion rate was reduced for 4 of the 97 exposed subjects (1 GAMUNEX, 3 GAMIMUNE N, 10%) on 4 occasions. Mild to moderate headache, nausea, and fever were the reported reasons. There were no anaphylactic or anaphylactoid reactions to GAMUNEX or GAMIMUNE N, 10%.
Any adverse events in trial 100176, irrespective of the causality assessment, reported by at least 5% of subjects during the 3-month trial are given in the following table.
| Adverse Event | GAMUNEX® | GAMIMUNE® N, 10% |
| No. of subjects: 48 | No. of subjects: 49 | |
| No. of subjects with AE | No. of subjects with AE | |
| (percentage of all subjects) | (percentage of all subjects) | |
| Headache | 28 (58%) | 30 (61%) |
| Ecchymosis, Purpura | 19 (40%) | 25 (51%) |
| Hemorrhage (All systems) | 14 (29%) | 16 (33%) |
| Epistaxis | 11 (23%) | 12 (24%) |
| Petechiae | 10 (21%) | 15 (31%) |
| Fever | 10 (21%) | 7 (14%) |
| Vomiting | 10 (21%) | 10 (20%) |
| Nausea | 10 (21%) | 7 (14%) |
| Thrombocytopenia | 7 (15%) | 8 (16%) |
| Accidental injury | 6 (13%) | 8 (16%) |
| Rhinitis | 6 (13%) | 6 (12%) |
| Pharyngitis | 5 (10%) | 5 (10%) |
| Rash | 5 (10%) | 6 (12%) |
| Pruritis | 4 (8%) | 1 (2%) |
| Asthenia | 3 (6%) | 5 (10%) |
| Abdominal Pain | 3 (6%) | 4 (8%) |
| Arthralgia | 3 (6%) | 6 (12%) |
| Back Pain | 3 (6%) | 3 (6%) |
| Dizziness | 3 (6%) | 3 (6%) |
| Flu Syndrome | 3 (6%) | 3 (6%) |
| Neck Pain | 3 (6%) | 1 (2%) |
| Anemia | 3 (6%) | 0 (0%) |
| Dyspepsia | 3 (6%) | 0 (0%) |
The subset of drug related adverse events in trial 100176 reported by at least 5% of subjects during the 3-month trial are given in the following table.
| Drug Related Adverse Event | GAMUNEX® | GAMIMUNE® N, 10% |
| No. of subjects: 48 | No. of subjects: 49 | |
| No. of subjects with drug related | No. of subjects with drug related | |
| AE (percentage of all subjects) | AE (percentage of all subjects) | |
| Headache | 24 (50%) | 24 (49%) |
| Vomiting | 6 (13%) | 8 (16%) |
| Fever | 5 (10%) | 5 (10%) |
| Nausea | 5 (10%) | 4 (8%) |
| Back Pain | 3 (6%) | 2 (4%) |
| Rash | 3 (6%) | 0 (0%) |
Serum samples were drawn to monitor the viral safety of the ITP subjects at baseline, nine days after the first infusion (for parvovirus B19), and 3 months after the first infusion of IGIV and at any time of premature discontinuation of the study. Viral markers of hepatitis C, hepatitis B, HIV-1, and parvovirus B19 were monitored by nucleic acid testing (NAT, PCR), and serological testing. There were no treatment related emergent findings of viral transmission for either GAMUNEX®, Immune Globulin Intravenous (Human), 10% Caprylate/Chromatography Purified, or GAMIMUNE® N, Immune Globulin Intravenous (Human), 10%.(17)
Treatment of Chronic Inflammatory Demyelinating Polyneuropathy
In study 100538, 113 subjects were exposed to GAMUNEX and 95 were exposed to Placebo. (See Clinical Studies [14.3]) As a result of the study design, the drug exposure with GAMUNEX was almost twice that of Placebo, with 1096 GAMUNEX infusions versus 575 Placebo infusions. Therefore, adverse reactions are reported per infusion (represented as frequency) to correct for differences in drug exposure between the 2 groups. The majority of loading-doses were administered over 2 days. The majority of maintenance-doses were administered over 1 day. Infusions were administered in the mean over 2.7 hours.
The following table shows the numbers of subjects per treatment group in the CIDP clinical trial, and the reason for discontinuation due to adverse events:
| Number of Subjects | Number of Subjects Discontinued due to Adverse Events | Adverse Event | |
| GAMUNEX® | 113 | 3 (2.7%) | Urticaria, Dyspnea, Bronchopneumonia |
| Placebo | 95 | 2 (2.1%) | Cerebrovascular Accident, Deep Vein Thrombosis |
Adverse events reported by at least 5% of subjects in any treatment group irrespective of causality are shown in the following table.
| MedDRA Preferred Term* | GAMUNEX®
No. of subjects: 113 | Placebo No. of subjects: 95 |
||||
| No. of Subjects (%) | No. of Adverse Events | Incidence density† | No. of Subjects (%) | No. of Adverse Events | Incidence density† |
|
| Any Adverse Event | 85 (75) | 377 | 0.344 | 45 (47) | 120 | 0.209 |
| Headache | 36 (32) | 57 | 0.052 | 8 (8) | 15 | 0.026 |
| Pyrexia (fever) | 15 (13) | 27 | 0.025 | 0 | 0 | 0 |
| Hypertension | 10 (9) | 20 | 0.018 | 4 (4) | 6 | 0.010 |
| Rash | 8 (7) | 13 | 0.012 | 1 (1) | 1 | 0.002 |
| Arthralgia | 8 (7) | 11 | 0.010 | 1 (1) | 1 | 0.002 |
| Asthenia | 9 (8) | 10 | 0.009 | 3 (3) | 4 | 0.007 |
| Chills | 9 (8) | 10 | 0.009 | 0 | 0 | 0 |
| Back pain | 9 (8) | 10 | 0.009 | 3 (3) | 3 | 0.005 |
| Nausea | 7 (6) | 9 | 0.008 | 3 (3) | 3 | 0.005 |
| Dizziness | 7 (6) | 3 | 0.006 | 1 (1) | 1 | 0.002 |
| Influenza | 6 (5) | 6 | 0.005 | 2 (2) | 2 | 0.003 |
Drug-related adverse events reported by at least 5% of subjects in any treatment group are reported in the table below. The most common drug-related events with GAMUNEX were headache and pyrexia:
| MedDRA Preferred Term* | GAMUNEX®
No. of subjects: 113 | Placebo No. of subjects: 95 |
||||
| No. of Subjects (%) | No. of Adverse Events | Incidence density† | No. of Subjects (%) | No. of Adverse Events | Incidence density† |
|
| Any drug-related | ||||||
| adverse event | 62 (55) | 194 | 0.177 | 16 (17) | 25 | 0.043 |
| Headache | 31 (27) | 44 | 0.040 | 6 (6) | 7 | 0.012 |
| Pyrexia (fever) | 15 (13) | 26 | 0.024 | 0 | 0 | 0 |
| Chills | 8 (7) | 9 | 0.008 | 0 | 0 | 0 |
| Hypertension | 7 (6) | 16 | 0.015 | 3 (3) | 3 | 0.005 |
| Rash | 6 (5) | 8 | 0.007 | 1 (1) | 1 | 0.002 |
| Nausea | 6 (5) | 7 | 0.006 | 3 (3) | 3 | 0.005 |
| Asthenia | 6 (5) | 6 | 0.005 | 0 | 0 | 0 |
Laboratory Abnormalities
During the course of the clinical program, ALT and AST elevations were identified in some subjects.
- For ALT, in the primary humoral immunodeficiency (PI) study (100175) treatment emergent elevations above the upper limit of normal were transient and observed among 14/80 (18%) of subjects in the GAMUNEX group versus 5/88 (6%) of subjects in the GAMIMUNE N, 10% group (p = 0.026).
- In the ITP study which employed a higher dose per infusion, but a maximum of only two infusions, the reverse finding was observed among 3/44 (7%) of subjects in the GAMUNEX group versus 8/43 (19%) of subjects in the GAMIMUNE N, 10% group (p = 0.118).
- In the CIDP study (100538), 15/113 (13%) of subjects in the GAMUNEX group and 7/95 (7%) in the Placebo group (p=0.168) had a treatment emergent transient elevation of ALT.
Elevations of ALT and AST were generally mild (<3 times upper limit of normal), transient, and were not associated with obvious symptoms of liver dysfunction.
GAMUNEX may contain low levels of anti-Blood Group A and B antibodies primarily of the IgG4 class. Direct antiglobulin tests (DAT or direct Coombs tests), which are carried out in some centers as a safety check prior to red blood cell transfusions, may become positive temporarily. Hemolytic events not associated with positive DAT findings were observed in clinical trials.(17, 30-33)
6.3 Postmarketing Experience
Because postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
GAMUNEX Postmarketing Experience
The following adverse reactions have been identified and reported during the post marketing use of GAMUNEX:
- Hematologic: Hemolytic anemia
- Infections and Infestations: Aseptic meningitis
General
The following adverse reactions have been identified and reported during the post marketing use of IGIV products (34):
- Respiratory: Apnea, Acute Respiratory Distress Syndrome (ARDS), TRALI, cyanosis, hypoxemia, pulmonary edema, dyspnea, bronchospasm
- Cardiovascular: Cardiac arrest, thromboembolism, vascular collapse, hypotension
- Neurological: Coma, loss of consciousness, seizures/convulsions, tremor
- Integumentary: Stevens-Johnson syndrome, epidermolysis, erythema multiforme, bullous dermatitis
- Hematologic: Pancytopenia, leukopenia, hemolysis, positive direct antiglobulin (Coombs test)
- General/Body as a Whole: Pyrexia, rigors
- Musculoskeletal: Back pain
- Gastrointestinal: Hepatic dysfunction, abdominal pain
7 DRUG INTERACTIONS
GAMUNEX may be diluted with 5% dextrose in water (D5/W). Admixtures of GAMUNEX with other drugs and intravenous solutions have not been evaluated. It is recommended that GAMUNEX be administered separately from other drugs or medications which the patient may be receiving. The product should not be mixed with IGIVs from other manufacturers.
The infusion line may be flushed before and after administration of GAMUNEX with 5% dextrose in water.
Various passively transferred antibodies in immunoglobulin preparations can confound the results of serological testing.
Antibodies in GAMUNEX may interfere with the response to live viral vaccines such as measles, mumps and rubella. Physicians should be informed of recent therapy with IGIVs, so that administration of live viral vaccines, if indicated, can be appropriately delayed 3 or more months from the time of IGIV administration. (See Patient Counseling Information [17])
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C. Animal reproduction studies have not been conducted with GAMUNEX. It is not known whether GAMUNEX can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. GAMUNEX should be given to a pregnant woman only if clearly needed.
8.4 Pediatric Use
Treatment of Primary Immunodeficiency
GAMUNEX was evaluated in 18 pediatric subjects (age range 0-16 years). Twenty-one percent of PI subjects (Study 100175) exposed to GAMUNEX were children. Pharmacokinetics, safety and efficacy were similar to those in adults with the exception that vomiting was more frequently reported in pediatrics (3 of 18 subjects). No pediatric-specific dose requirements were necessary to achieve serum IgG levels.
One subject, a 10-year-old boy, died suddenly from myocarditis 50 days after his second infusion of GAMUNEX. The death was judged to be unrelated to GAMUNEX.
Treatment of Idiopathic Thrombocytopenic Purpura
GAMUNEX was evaluated in 12 pediatric subjects with acute ITP. Twenty-five percent of the acute ITP subjects (Study 100176) exposed to GAMUNEX were children. Pharmacokinetics, safety and efficacy were similar to those in adults with the exception that fever was more frequently reported in pediatrics (6 of 12 subjects). No pediatric-specific dose requirements were necessary to achieve serum IgG levels.
8.5 Geriatric Use
Patients > 65 years of age may be at increased risk for developing certain adverse reactions such as thromboembolic events and acute renal failure. (See Boxed Warning, Warnings and Precautions [5.2]) Clinical studies of GAMUNEX did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
| Clinical Study | Indication | Number of Subjects | |
| < 65 years | ≥ 65 years | ||
| 100175 | PI | 78 | 9 |
| 100152 | PI | 18 | 0 |
| 100174 | PI | 20 | 0 |
| 10039 | PI | 19 | 0 |
| 100213 | ITP | 22 | 6 |
| 100176 | ITP | 44 | 4 |
| 10038 | ITP | 18 | 3 |
| 100538 | CIDP | 44 | 15 |
11 DESCRIPTION
GAMUNEX is a ready-to-use sterile solution of human immune globulin protein for intravenous administration. GAMUNEX consists of 9%–11% protein in 0.16–0.24 M glycine. Not less than 98% of the protein has the electrophoretic mobility of gamma globulin. GAMUNEX contains trace levels of fragments, IgA (average 0.046 mg/mL), and IgM. The distribution of IgG subclasses is similar to that found in normal serum. GAMUNEX doses of 1 g/kg correspond to a glycine dose of 0.15 g/kg. While toxic effects of glycine administration have been reported,(35) the doses and rates of administration were 3 – 4 fold greater than those for GAMUNEX. In another study, it was demonstrated that intravenous bolus doses of 0.44 g/kg glycine were not associated with serious adverse effects.(36) Caprylate is a saturated medium-chain (C8) fatty acid of plant origin. Medium chain fatty acids are considered to be essentially non-toxic. Human subjects receiving medium chain fatty acids parenterally have tolerated doses of 3.0 to 9.0 g/kg/day for periods of several months without adverse effects.(37) Residual caprylate concentrations in the final container are no more than 0.216 g/L (1.3 mmol/L).The measured buffer capacity is 35 mEq/L and the osmolality is 258 mOsmol/kg solvent, which is close to physiological osmolality (285-295 mOsmol/kg). The pH of GAMUNEX is 4.0 – 4.5. GAMUNEX contains no preservative and is latex-free.
GAMUNEX is made from large pools of human plasma by a combination of cold ethanol fractionation, caprylate precipitation and filtration, and anion-exchange chromatography. Isotonicity is achieved by the addition of glycine. GAMUNEX is incubated in the final container (at the low pH of 4.0 – 4.3), for a minimum of 21 days at 23° to 27°C. The product is intended for intravenous administration.
The capacity of the manufacturing process to remove and/or inactivate enveloped and non-enveloped viruses has been validated by laboratory spiking studies on a scaled down process model, using the following enveloped and non-enveloped viruses: human immunodeficiency virus, type I (HIV-1) as the relevant virus for HIV-1 and HIV–2; bovine viral diarrhea virus (BVDV) as a model for hepatitis C virus; pseudorabies virus (PRV) as a model for large DNA viruses (e.g., herpes viruses); Reo virus type 3 (Reo) as a model for non-enveloped viruses and for its resistance to physical and chemical inactivation; hepatitis A virus (HAV) as relevant non-enveloped virus, and porcine parvovirus (PPV) as a model for human parvovirus B19.
Overall virus reduction was calculated only from steps that were mechanistically independent from each other and truly additive. In addition, each step was verified to provide robust virus reduction across the production range for key operating parameters.
|
||||||
| Process Step | Log 10 Virus Reduction | |||||
| Enveloped Viruses | Non-enveloped Viruses | |||||
| HIV | PRV | BVDV | Reo | HAV | PPV | |
| Caprylate Precipitation/Depth Filtration | C/I * | C/I | 2.7 | ≥ 3.5 | ≥ 3.6 | 4.0 |
| Caprylate Incubation | ≥ 4.5 | ≥ 4.6 | ≥ 4.5 | NA† | NA | NA |
| Depth Filtration‡ | CAP§ | CAP | CAP | ≥ 4.3 | ≥ 2.0 | 3.3 |
| Column Chromatography | ≥ 3.0 | ≥ 3.3 | 4.0 | ≥ 4.0 | ≥ 1.4 | 4.2 |
| Low pH Incubation (21 days) | ≥ 6.5 | ≥ 4.3 | ≥ 5.1 | NA | NA | NA |
| Global Reduction | ≥ 14.0 | ≥ 12.2 | ≥ 16.3 | ≥ 7.5 | ≥ 5.0 | 8.2 |
Additionally, the manufacturing process was investigated for its capacity to decrease the infectivity of an experimental agent of transmissible spongiform encephalopathy (TSE), considered as a model for the vCJD and CJD agents.(38-42)
Several of the individual production steps in the GAMUNEX manufacturing process have been shown to decrease TSE infectivity of that experimental model agent. TSE reduction steps include two depth filtrations (in sequence, a total of ≥ 6.6 logs). These studies provide reasonable assurance that low levels of CJD/vCJD agent infectivity, if present in the starting material, would be removed.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Treatment of Primary Humoral Immunodeficiency
GAMUNEX supplies a broad spectrum of opsonic and neutralizing IgG antibodies against bacteria or their toxins. The mechanism of action in PI has not been fully elucidated.
12.3 Pharmacokinetics
Two randomized pharmacokinetic crossover trials were carried out with GAMUNEX in 38 subjects with Primary Humoral Immunodeficiencies given 3 infusions 3 or 4 weeks apart of test product at a dose of 100-600 mg/kg body weight per infusion. One trial compared the pharmacokinetic characteristics of GAMUNEX to GAMIMUNE N, 10% (study 100152), and the other trial compared the pharmacokinetics of GAMUNEX (10% strength) with a 5% concentration of this product (study 100174). The ratio of the geometric least square means for dose-normalized IgG peak levels of GAMUNEX and GAMIMUNE N, 10% was 0.996. The corresponding value for the dose-normalized area under the curve (AUC) of IgG levels was 0.990. The results of both PK parameters were within the pre-established limits of 0.080 and 1.25. Similar results were obtained in the comparison of GAMUNEX 10% to a 5% concentration of GAMUNEX.(31,32)
The main pharmacokinetic parameters of GAMUNEX, measured as total IgG in study 100152 are displayed below:
| GAMUNEX® | GAMIMUNE® N, 10% | |||||||
| N | Mean | SD | Median | N | Mean | SD | Median | |
| Cmax (mg/mL) | 17 | 19.04 | 3.06 | 19.71 | 17 | 19.31 | 4.17 | 19.30 |
| Cmax-norm (kg/mL) | 17 | 0.047 | 0.007 | 0.046 | 17 | 0.047 | 0.008 | 0.047 |
| AUC(0-tn)* | 17 | 6746.48 | 1348.13 | 6949.47 | 17 | 6854.17 | 1425.08 | 7119.86 |
| AUC(0-tn) norm* (kg*hr/mL) | 17 | 16.51 | 1.83 | 16.95 | 17 | 16.69 | 2.04 | 16.99 |
| T 1/2†
(days) | 16 | 35.74 | 8.69 | 33.09 | 16 | 34.27 | 9.28 | 31.88 |
The two pharmacokinetic trials with GAMUNEX show the IgG concentration/time curve follows a biphasic slope with a distribution phase of about 5 days characterized by a fall in serum IgG levels to about 65-75% of the peak levels achieved immediately post-infusion. This phase is followed by the elimination phase with a half-life of approximately 35 days.(31, 32) IgG trough levels were measured over nine months in the therapeutic equivalence trial (100175). Mean trough levels were 7.8 +/- 1.9 mg/mL for the GAMUNEX treatment group and 8.2 +/- 2.0 mg/mL for the GAMIMUNE N, 10% control group.(30)
14 CLINICAL STUDIES
14.1 Treatment of Primary Humoral Immunodeficiency
In a randomized, double-blind, parallel group clinical trial with 172 subjects with primary humoral immunodeficiencies (study 100175) GAMUNEX was demonstrated to be at least as efficacious as GAMIMUNE N, 10% in the prevention of any infection, i.e. validated plus clinically defined, non-validated infections of any organ system, during a nine month treatment period. Twenty six subjects were excluded from the Per Protocol analysis (2 due to non-compliance and 24 due to protocol violations). The endpoint was the proportion of subjects with at least one of the following validated infections: pneumonia, acute sinusitis and acute exacerbations of chronic sinusitis.
| GAMUNEX® | GAMIMUNE® N, 10% | Mean Difference | p-Value | |
| (n=73) | (n=73) | (90% confidence interval) | ||
| No. of subjects with | No. of subjects with | |||
| at least one infection | at least one infection | |||
| Validated Infections | 9 (12%) | 17 (23%) | -0.117 | 0.06 |
| (-0.220, -0.015) | ||||
| Acute Sinusitis | 4 (5%) | 10 (14%) | ||
| Exacerbation of | ||||
| Chronic Sinusitis | 5 (7%) | 6 (8%) | ||
| Pneumonia | 0 (0%) | 2 (3%) | ||
| Any Infection | 56 (77%) | 57 (78%) | -0.020 | 0.78 |
| (Validated plus | (-0.135, 0.096) | |||
| Clinically defined | ||||
| non-validated Infections) |
The annual rate of validated infections (Number of Infections/year/subject) was 0.18 in the group treated with GAMUNEX and 0.43 in the group treated with GAMIMUNE N, 10% (p=0.023). The annual rates for any infection (validated plus clinically-defined, non-validated infections of any organ system) were 2.88 and 3.38, respectively (p=0.300).(30, 43)
14.2 Treatment of Idiopathic Thrombocytopenic Purpura
A double-blind, randomized, parallel group clinical trial with 97 ITP subjects was carried out to prove the hypothesis that GAMUNEX was at least as effective as GAMIMUNE N, 10% in raising platelet counts from less than or equal to 20 ×109/L to more than 50 ×109/L within 7 days after treatment with 2 g/kg IGIV (study 100176). Twenty-four percent of the subjects were less than or equal to 16 years of age.
GAMUNEX was demonstrated to be at least as effective as GAMIMUNE N, 10% in the treatment of adults and children with acute or chronic ITP.(11)
| GAMUNEX® | GAMIMUNE® N, 10% | Mean Difference | |
| (n=39) | (n=42) | (90% confidence interval) | |
| By Day 7 | 35 (90%) | 35 (83%) | 0.075 |
| (-0.037, 0.186) | |||
| By Day 23 | 35 (90%) | 36 (86%) | 0.051 |
| (-0.058, 0.160) | |||
| Sustained for 7 days | 29 (74%) | 25 (60%) | 0.164 |
| (0.003, 0.330) |
A trial was conducted to evaluate the clinical response to rapid infusion of GAMUNEX in patients with ITP. The study involved 28 chronic ITP subjects, wherein the subjects received 1 g/kg GAMUNEX on three occasions for treatment of relapses. The infusion rate was randomly assigned to 0.08, 0.11, or 0.14 mL/kg/min (8, 11 or 14 mg/kg/min). Pre-medication with corticosteroids to alleviate infusion-related intolerability was not permitted. Pre-treatment with antihistamines, anti-pyretics and analgesics was permitted. The average dose was approximately 1 g/kg body weight at all three prescribed rates of infusion (0.08, 0.11 and 0.14 mL/kg/min). All patients were administered each of the three planned infusions except seven subjects. Based on 21 patients per treatment group, the a posteriori power to detect twice as many drug-related adverse events between groups was 23%. Of the seven subjects that did not complete the study, five did not require additional treatment, one withdrew because he refused to participate without concomitant medication (prednisone) and one experienced an adverse event (hives); however, this was at the lowest dose rate level (0.08 mL/kg/min).
14.3 Treatment of Chronic Inflammatory Demyelinating Polyneuropathy
A multi-center, randomized, double-blind Placebo-controlled trial (study 100538, The Immune Globulin Intravenous (Human), 10% Caprylate/Chromatography Purified CIDP Efficacy or ICE study) was conducted with GAMUNEX.(44) This study included two separately randomized study periods to assess whether GAMUNEX was more effective than Placebo for the treatment of CIDP (assessed in the Efficacy Period for up to 24 weeks) and whether long-term administration of GAMUNEX could maintain long-term benefit (assessed in the 24 week Randomized Withdrawal Period).
In the Efficacy Period, there was a requirement for Rescue (crossover) to the alternate study drug if the subject did not improve and maintain this improvement until the end of the 24 week treatment period. Subjects entering the Rescue phase followed the same dosing and schedule as in the Efficacy period. Any subject who was Rescued (crossed over) and did not improve and maintain this improvement was withdrawn from the study.
Subjects who completed 24 weeks treatment in the Efficacy period or Rescue phase and responded to therapy were eligible for entry into a double-blind Randomized Withdrawal Period. Eligible subjects were re-randomized to GAMUNEX or Placebo. Any subject who relapsed was withdrawn from the study.
The Efficacy Period and the Rescue treatment started with a loading dose of 2 g/kg bw of GAMUNEX or equal volume of Placebo given over 2-4 consecutive days. All other infusions (including the first infusion of the Randomized Withdrawal Period) were given as maintenance doses of 1 g/kg bw (or equivalent volume of Placebo) every three weeks.
The Responder rates of the GAMUNEX and Placebo treatment groups was measured by the INCAT score. The INCAT (Inflammatory Neuropathy Cause and Treatment) scale is used to assess functional disability of both upper and lower extremities in demyelinating polyneuropathy. The INCAT scale has upper and lower extremity components (maximum of 5 points for upper (arm disability) and maximum of 5 points for lower (leg disability)) that add up to a maximum of 10-points (0 is normal and 10 is severely incapacitated).(45) At the start of the efficacy portion of the study, the INCAT scores were as follows: Upper Extremity mean was 2.2 ± 1.0, and median was 2.0 with a range of 0 to 5; Lower Extremity mean was 1.9 ± 0.9, and median was 2.0 with a range of 1 to 5; Total Overall Score mean was 4.2 ± 1.4, and median was 4.0 with a range of 2 to 9. A Responder was defined as a subject with at least 1-point improvement from baseline in the adjusted INCAT score that was maintained through 24 weeks.
Significantly more subjects with CIDP responded to GAMUNEX: 28 of 59 subjects (47.5%) responded to GAMUNEX compared with 13 of 58 subjects (22.4%) administered Placebo (25% difference; [95% CI: 7%-43%]; p=0.006). The study included both subjects who were IGIV naive and subjects who had previous IGIV experience. The outcome was influenced by the group of subjects who experienced prior therapy with IGIV, as shown by the outcomes table, below.
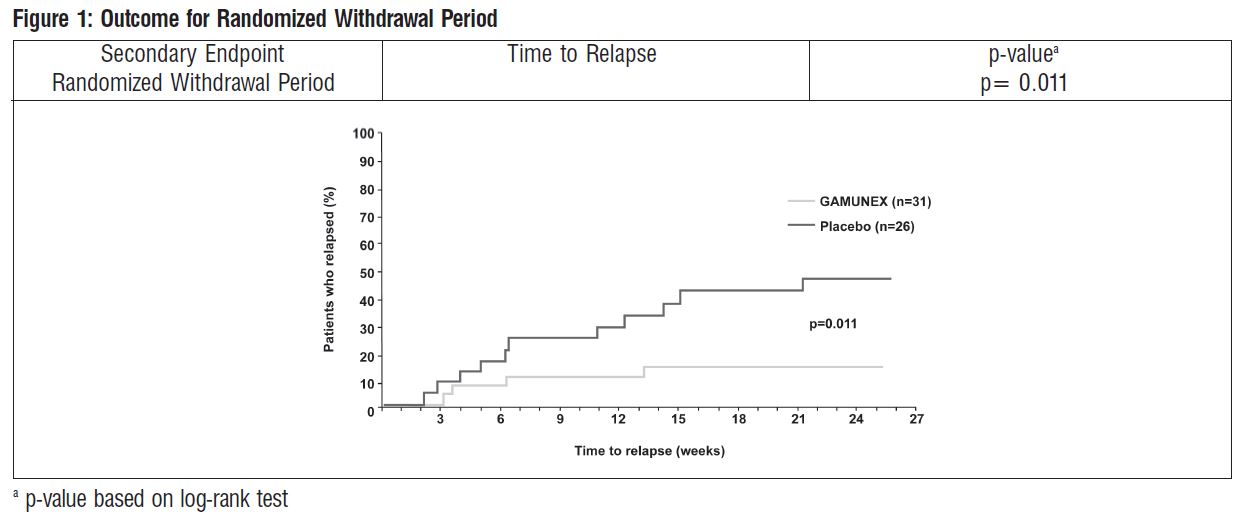
Time to relapse for the subset of 57 subjects who previously responded to GAMUNEX was evaluated: 31 were randomly reassigned to continue to receive GAMUNEX and 26 subjects were randomly reassigned to Placebo in the Randomized Withdrawal Period. Subjects who continued to receive GAMUNEX experienced a significantly longer time to relapse versus subjects treated with Placebo (p=0.011). The probability of relapse was 13% with GAMUNEX versus 45% with Placebo (hazard ratio, 0.19 [95% confidence interval, 0.05, 0.70]).
|
|||||
| Efficacy Period | GAMUNEX® | Placebo | p-value* | ||
| Responder | Non-Responder | Responder | Non-Responder | ||
| All Subjects | 28/59 (47.5%) | 31/59 (52.5%) | 13/58 (22.4%) | 45/58 (77.6%) | 0.006 |
| IGIV - Naïve Subjects | 17/39 (43.6%) | 22/39 (56.4%) | 13/46 (28.3%) | 33/46 (71.7%) | 0.174 |
| IGIV - Experienced Subjects | 11/20 (55.0%) | 9/20 (45.0%) | 0/12 (0%) | 12/12 (100%) | 0.002 |
The following table shows outcomes for the Rescue Phase (which are supportive data):
|
|||||
| Rescue Phase | GAMUNEX® | Placebo | p-value* | ||
| Success | Failure | Success | Failure | ||
| All Subjects | 25/45 (55.6%) | 20/45 (44.4%) | 6/23 (26.1%) | 17/23 (73.9%) | 0.038 |
| IGIV - Naïve Subjects | 19/33(57.6%) | 14/33 (42.4%) | 6/18 (33.3%) | 12/18 (66.7%) | 0.144 |
| IGIV - Experienced Subjects | 6/12 (50%) | 6/12 (50%) | 0/5 (0%) | 5/5(100%) | 0.102 |
The following Kaplan-Meier curves show the outcomes for the Randomized Withdrawal Period:
15 REFERENCES
- Cayco AV, Perazella MA, Hayslett J P. Renal insufficiency after intravenous immune globulin therapy: a report of two cases and an analysis of the literature. J Am Soc Nephrol, 1997. 8(11): p. 1788-94.
- Ammann AJ, et al. Use of intravenous gamma-globulin in antibody immunodeficiency: results of a multicenter controlled trial. Clin Immunol Immunopathol, 1982. 22(1): p. 60-7.
- Buckley RH, Schiff RI. The use of intravenous immune globulin in immunodeficiency diseases. N Engl J Med, 1991. 325(2): p. 110-7.
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol, 1999. 92(1): p. 34-48.
- Nolte M T, et al. Intravenous immunoglobulin therapy for antibody deficiency. Clin Exp Immunol, 1979. 36(2): p. 237-43.
- Pruzanski W, et al. Relationship of the dose of intravenous gammaglobulin to the prevention of infections in adults with common variable immunodeficiency. Inflammation, 1996. 20(4): p. 353-9.
- Roifman CM, Levison H, Gelfand EW. High-dose versus low-dose intravenous immunoglobulin in hypogammaglobulinaemia and chronic lung disease. Lancet, 1987. 1(8541): p. 1075-7.
- Sorensen RU, Polmar SH. Efficacy and safety of high-dose intravenous immune globulin therapy for antibody deficiency syndromes. Am J Med, 1984. 76(3A): p. 83-90.
- Stephan JL, et al. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr, 1993. 123(4): p. 564-72.
- Blanchette VS, Kirby MA, and Turner C. Role of intravenous immunoglobulin G in autoimmune hematologic disorders. Semin Hematol, 1992. 29(3 Suppl 2): p. 72-82.
- Lazarus AH, Freedman J, Semple JW. Intravenous immunoglobulin and anti-D in idiopathic thrombocytopenic purpura (ITP): mechanisms of action. Transfus Sci, 1998. 19(3): p. 289-94.
- Semple JW, Lazarus AH, Freedman J. The cellular immunology associated with autoimmune thrombocytopenic purpura: an update. Transfus Sci, 1998. 19(3): p. 245-51.
- Imbach PA. Harmful and beneficial antibodies in immune thrombocytopenic purpura. Clin Exp Immunol, 1994. 97(Suppl 1): p. 25-30.
- Bussel JB. Fc receptor blockade and immune thrombocytopenic purpura. Semin Hematol, 2000. 37(3): p. 261-6.
- Imbach P, et al. Immunthrombocytopenic purpura as a model for pathogenesis and treatment of auto immunity. Eur J Pediatr, 1995. 154(9 Suppl 4): p. S60-4.
- Guyton A. Textbook of Medical Physiology. 5th Edition. 1976, Philadelphia: W.B. Saunders. 499-500.
- Cyrus P, F.G., Kelleher J, Schwartz L. A Randomized, Double-Blind, Multicenter, Parallel Group Trial Comparing the Safety, and Efficacy of IGIV-Chromatography, 10% (Experimental) with IGIV-Solvent Detergent Treated, 10% (Control) in Patients with Idiopathic (Immune) Thrombocytopenic Purpura (ITP), 2000. Report on file.
- Steinberger BA, Ford SM, Coleman TA. Intravenous Immunoglobulin Therapy Results in Post-infusional Hyperproteinemia, Increased Serum Viscosity, and Pseudohyponatremia. Am J Hematol 73:97-100 (2003).
- Dalakas MC. High-dose intravenous Immunoglobulin and serum viscosity: risk of precipitating thromboembolic events. Neurology, 44:223-226.
- Woodruff RK, Grigg A P, Firkin FC, Smith IL. Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet 1986;2:217-218.
- Wolberg AS, Kon RH, Monroe DM, Hoffman M. Coagulation factor XI is a contaminant in intravenous immunoglobulin preparations. Am J Hematol 2000; 65,30-34.
- Casteels-Van Daele M, et al. Intravenous immune globulin and acute aseptic meningitis [letter]. N Engl J Med, 1990. 323(9): p. 614-5.
- Kato E, et al. Administration of immune globulin associated with aseptic meningitis [letter]. Jama, 1988. 259(22): p. 3269-71.
- Scribner CL, et al. Aseptic meningitis and intravenous immunoglobulin therapy [editorial; comment]. Ann Intern Med, 1994. 121(4): p. 305-6.
- Copelan EA, Strohm PL, Kennedy MS, Tutschka PJ. Hemolysis following intravenous immune globulin therapy. Transfusion 1986;26: 410 - 41 2 .
- Thomas MJ, Misbah SA, Chapel HM, Jones M, Elrington G, Newsom-Davis J. Hemolysis after high-dose intravenous Ig. Blood 1993;15:3789.
- Wilson JR, Bhoopalam N, Fisher M. Hemolytic anemia associated with intravenous immunoglobulin. Muscle & Nerve 1997;20:1142-1145.
- Kessary-Shoham H, Levy Y, Shoenfeld Y, Lorber M, Gershon H. In vivo administration of intravenous immunoglobulin (IVIg) can lead to enhanced erythrocyte sequestration. J Autoimmune 1999;13:129-135.
- Rizk A, Gorson KC, Kenney L, Weinstein R. Transfusion-related acute lung injury after the infusion of IVIG. Transfusion 2001:41:264-268.
- Kelleher J, F.G., Cyrus P, Schwartz L. A Randomized, Double-Blind, Multicenter, Parallel Group Trial Comparing the Safety and Efficacy of IGIV-Chromatography, 10% (Experimental) with IGIV-Solvent Detergent Treated, 10% (Control) in Patients with Primary Immune Deficiency (PID), 2000. Report on file.
- Bayever E, M.F., Sundaresan P, Collins S. Randomized, Double-Blind, Multicenter, Repeat Dosing, Cross-Over Trial Comparing the Safety, Pharmacokinetics, and Clinical Outcomes of IGIV-Chromatography, 10% (Experimental) with IGIV-Solvent Detergent Treated, 10% (Control) in Patients with Primary Humoral Immune Deficiency (BAY-41-1000-100152). MMRR-1512/1, 1999.
- Lathia C, E.B., Sundaresan PR, Schwartz L. A Randomized, Open-Label, Multicenter, Repeat Dosing, Cross-Over Trial Comparing the Safety, Pharmacokinetics, and Clinical Outcomes of IGIV-Chromatography, 5% with IGIV-Chromatography 10% in Patients with Primary Humoral Immune Deficiency (BAY-41-1000-100174). 2000.
- Kelleher J, S.L. IGIV-C 10% Rapid Infusion Trial in Idiopathic (Immune) Thrombocytopenic Purpura (ITP), 2001. Report on file.
- Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin. Trans Med Rev 2003; 17,241-251.
- Hahn RG, Stalberg H P, Gustafsson SA. Intravenous infusion of irrigating fluids containing glycine or mannitol with and without ethanol. J Urol, 1989. 142(4): p. 1102-5.
- Ta i VM, M.E., Lee-Brotherton V, Manley JJ, Nestmann ER, Daniels JM. Safety Evaluation of Intravenous Glycine in Formulation Development. in J Pharm Pharmaceut Sci. 2000.
- Traul KA, et al. Review of the toxicologic properties of medium-chain triglycerides. Food Chem Toxicol, 2000. 38(1): p. 79-98.
- Stenland CJ, Lee DC, Brown P, et al. Partitioning of human and sheep forms of the pathogenic prion protein during the purification of therapeutic proteins from human plasma. Transfusion 2002. 42(11):1497-500.
- Lee DC, Stenland CJ, Miller JL, et al. A direct relationship between the partitioning of the pathogenic prion protein and transmissible spongiform encephalopathy infectivity during the purification of plasma proteins. Transfusion 2001. 41(4):449-55.
- Lee DC, Stenland CJ, Hartwell RC, et al. Monitoring plasma processing steps with a sensitive Western blot assay for the detection of the prion protein. J Virol Methods 2000. 84(1):77-89.
- Cai K, Miller JL, Stenland CJ, et al. Solvent-dependent precipitation of prion protein. Biochim Biophys Acta 2002. 1597(1):28-35.
- Trejo SR, Hotta JA, Lebing W, et al. Evaluation of virus and prion reduction in a new intravenous immunoglobulin manufacturing process. Vox Sang 2003. 84(3):176-87.
- Data on File.
- Hughes RAC, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate/chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomized Placebo-controlled trial. Lancet Neurol 2008. 7:136-144.
- Hughes R, Bensa S, Willison H, Van den B P, Comi G, Illa I, et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol 2001 Aug;50(2):195-201.
16 HOW SUPPLIED/STORAGE AND HANDLING
GAMUNEX is supplied in single-use, tamper evident vials (shrink band) containing the labeled amount of functionally active IgG. The three larger vial size labels incorporate integrated hangers. The components used in the packaging for GAMUNEX are latex-free. GAMUNEX is supplied in the following sizes:
| NDC Number | Size | Grams Protein |
| 13533-645-12 | 10 mL | 1.0 |
| 13533-645-15 | 25 mL | 2.5 |
| 13533-645-20 | 50 mL | 5.0 |
| 13533-645-71 | 100 mL | 10.0 |
| 13533-645-24 | 200 mL | 20.0 |
GAMUNEX may be stored for 36 months at 2 - 8°C (36 - 46°F), AND product may be stored at temperatures not to exceed 25°C (77°F) for up to 6 months anytime during the 36 month shelf life, after which the product must be immediately used or discarded. Do not freeze. Do not use after expiration date.
17 PATIENT COUNSELING INFORMATION
(See Boxed Warning and Warnings and Precautions Sections)
Inform patients to immediately report the following to their physician:
- signs and symptoms of renal failures, such as decreased urine output, sudden weight gain, fluid retention/edema, and/or shortness of breath
- signs and symptoms of aseptic meningitis, such as headache, neck stiffness, drowsiness, fever, sensitivity to light, painful eye movements, nausea, and vomiting
- signs and symptoms of hemolysis, such as fatigue, increased heart rate, yellowing of the skin or eyes, and dark-colored urine
- signs and symptoms of TRALI, such as severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. TRALI typically occurs within 1 to 6 hours following transfusion.
Inform patients that GAMUNEX is made from human plasma and may contain infectious agents that can cause disease (e.g., viruses, and, theoretically, the CJD agent). Inform patients that the risk GAMUNEX may transmit an infectious agent has been reduced by screening plasma donors for prior exposure to certain viruses, by testing the donated plasma for certain virus infections and by inactivating and/or removing certain viruses during manufacturing.
Inform patients that administration of IgG may interfere with the response to live viral vaccines such as measles, mumps and rubella. Inform patients to notify their immunizing physician of therapy with GAMUNEX.
Manufactured by:
Rx only
Talecris Biotherapeutics, Inc.
Research Triangle Park, NC 27709 USA
U.S. License No. 1716
08939392/08939393
October 2008
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
NDC 13533-645-20
Immune Globulin
Intravenous (Human), 10%
gamunex™
Caprylae/Chromatography Purified
Solution for Infusion
5 Grams Protein in 50 ml
Talecris Biotherapeutics, Inc.
Research Triangle Park,
NC 27709 USA
U.S. License No. 1716
The patient and physician should discuss the risks and benefits of this product.
FOR INTRAVENOUS USE ONLY.
Refer to package insert for dosage and administration and compatibility with other solutions.
Gamunex may be stored for 36 months at 2–8°C (36–46°F), AND product may be stored at temperatures not to exceed 25°C (77°F) for up to 6 months anytime during the 36 month shelf life, after which the product must be immediately used or discarded.
Single Dose Vial
Do not use if turbid.
Rx only
Gamunex Lot
Lot Exp.

Immune Globulin Intravenous (Human), 10%
gamunex™
Caprylate/Chromatography Purified
Solution for Infusion
NDC 13533-645-20
5 Grams Protein in 50 ml
Talecris Biotherapeutics
Contents: One bottle of Gamunex
No preservative
Sterile, Non-pyrogenic
Gamunex may be stored for 36 months at 2–8°C (36–46°F), AND product may be stored at temperatures not to exceed 25°C (77°F) for up to 6 months anytime during the 36 month shelf life, after which the product must be immediately used or discarded.
Date removed from refrigeration:
The patient and physician should discuss the risks and benefits of this product.
FOR INTRAVENOUS USE ONLY.
Refer to package insert for dosage and administration and compatibility with other solutions.
Single Dose Vial
Do not use if turbid.
Discard unused portion. Do not store after entry into bottle.
Contains no preservative
Do not freeze.
Each milliliter (mL) contains approximately 100 mg of protein, not less than 98% of which is immunoglobulin, and approximately 15 mg glycine.
If the shrink band is absent or shows any sign of tampering, do not use the product and notify Talecris Biotherapeutics, Inc. immediately.
Not Returnable for Credit or Exchange
Rx Only
08908160
Talecris Biotherapeutics, Inc.
Research Triangle Park,
NC 27709 USA
U.S. License No. 1716

| GAMUNEX
immune globulin intravenous (human), 10% caprylate chromatography purified injection |
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
| Labeler - GRIFOLS USA, LLC (048987452) |