INCRELEX
-
mecasermin injection, solution
Tercica, Inc.
----------
INCRELEX® (mecasermin [rDNA origin] injection)DESCRIPTION
INCRELEX® (mecasermin [rDNA origin] injection) is an aqueous solution for injection containing human insulin-like growth factor-1 (rhIGF-1) produced by recombinant DNA technology. IGF-1 consists of 70 amino acids in a single chain with three intramolecular disulfide bridges and a molecular weight of 7649 daltons. The amino acid sequence of the product is identical to that of endogenous human IGF-1. The rhIGF-1 protein is synthesized in bacteria (E. coli) that have been modified by the addition of the gene for human IGF-1.
Primary Amino Acid Sequence of rhIGF-1
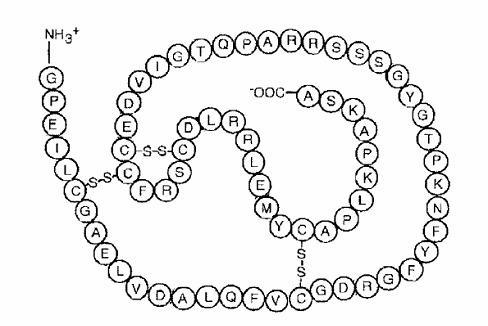
INCRELEX® is a purified preparation. Biological potency is determined using a bioassay.
INCRELEX® is a sterile, aqueous, clear and colorless solution intended for subcutaneous injection. Each multi-dose vial of INCRELEX® contains 10 mg/mL mecasermin, 9 mg/mL benzyl alcohol, 5.84 mg/mL sodium chloride, 2 mg/mL polysorbate 20, and 0.05M acetate at a pH of approximately 5.4.
CLINICAL PHARMACOLOGY
General
Insulin-like growth factor-1 (IGF-1) is the principal hormonal mediator of statural growth. Under normal circumstances, growth hormone (GH) binds to its receptor in the liver, and other tissues, and stimulates the synthesis/secretion of IGF-1. In target tissues, the Type 1 IGF-1 receptor, which is homologous to the insulin receptor, is activated by IGF-1, leading to intracellular signaling which stimulates multiple processes leading to statural growth. The metabolic actions of IGF-1 are in part directed at stimulating the uptake of glucose, fatty acids, and amino acids so that metabolism supports growing tissues.
The following actions have been demonstrated for endogenous human IGF-1:
Tissue Growth – 1) Skeletal growth occurs at the cartilage growth plates of the epiphyses of bones where stem cells divide to produce new cartilage cells or chondrocytes. The growth of chondrocytes is under the control of IGF-1 and GH. The chondrocytes become calcified so that new bone is formed allowing the length of the bones to increase. This results in skeletal growth until the cartilage growth plates fuse at the end of puberty. 2) Cell growth: IGF-1 receptors are present on most types of cells and tissues. IGF-1 has mitogenic activities that lead to an increased number of cells in the body. 3) Organ growth: Treatment of IGF-1 deficient rats with rhIGF-1 results in whole body and organ growth.
Carbohydrate Metabolism –IGF-1 suppresses hepatic glucose production and stimulates peripheral glucose utilization and therefore has a hypoglycemic potential. IGF-1 has inhibitory effects on insulin secretion.
Pharmacokinetics
Absorption – While the bioavailability of rhIGF-1 after subcutaneous administration in healthy subjects has been reported to be close to 100%, the absolute bioavailability of INCRELEX® given subcutaneously to subjects with primary insulin-like growth factor-1 deficiency (Primary IGFD) has not been determined.
Distribution – In blood, IGF-1 is bound to six IGF binding proteins, with > 80% bound as a complex with IGFBP-3 and an acid-labile subunit. IGFBP-3 is greatly reduced in subjects with severe Primary IGFD, resulting in increased clearance of IGF-1 in these subjects relative to healthy subjects. The total IGF-1 volume of distribution after subcutaneous administration in subjects with severe Primary IGFD is estimated to be 0.257 (± 0.073) L/kg at an INCRELEX® dose of 0.045 mg/kg, and is estimated to increase as the dose of INCRELEX® increases.
Metabolism – Both the liver and the kidney have been shown to metabolize IGF-1.
Excretion – The mean terminal t1/2 after single subcutaneous administration of 0.12 mg/kg INCRELEX® in pediatric subjects with severe Primary IGFD is estimated to be 5.8 hours. Clearance of INCRELEX® is inversely proportional to IGF binding protein-3 (IGFBP-3) levels and CL/F is estimated to be 0.04 L/hr/kg at 3 mcg/mL IGFBP-3.
| Cmax | Tmax | AUC0-8 | t1/2 | Vd/F | CL/F | |
| (ng/mL) | (hr) | (hr*ng/mL) | (hr) | (L/kg) | (L/hr/kg) | |
| Cmax = maximum concentration; Tmax = time of maximum concentration; AUC0-8 = area under the curve; t1/2 = half-life; Vd/F = volume of distribution; CL/F = systemic clearance; SC = subcutaneous injection; CV% = coefficient of variation in %. | ||||||
| Male/female data combined, ages 12 to 22 years. | ||||||
| a Data represents 3 subjects each at doses 0.015, 0.03, 0.06, and 0.12 mg/kg SC. | ||||||
| PK parameters based on baseline adjusted plasma concentrations. | ||||||
| n | 3 | 3 | 3 | 3 | 12a | 12a |
| Mean | 234 | 2 | 2932 | 5.8 | 0.257 | 0.0424 |
| CV% | 23 | 0 | 50 | 64 | 28 | 38 |
 |
Special Populations
Geriatric – The pharmacokinetics of INCRELEX® have not been studied in subjects greater than 65 years of age.
Gender – In children with Primary IGFD and in healthy adults there were no apparent differences between males and females in the pharmacokinetics of INCRELEX®.
Race – No information is available.
Renal insufficiency – No studies have been conducted in Primary IGFD children with renal impairment.
Hepatic insufficiency – No studies have been conducted to determine the effect of hepatic impairment on the pharmacokinetics of rhIGF-1.
CLINICAL TRIALS
Effects of INCRELEX® Treatment in Children with Severe Primary Insulin-like Growth Factor-1 Deficiency (Primary IGFD)
Five clinical studies (four open-label and one double-blind, placebo-controlled), with subcutaneous (SC) doses of INCRELEX® generally ranging from 0.06 to 0.12 mg/kg (60 to 120 μg/kg) administered twice daily (BID), were conducted in 71 pediatric subjects with severe Primary IGFD. Patients were enrolled in the trials on the basis of extreme short stature, slow growth rates, low IGF-1 serum concentrations, and normal growth hormone secretion. Data from these 5 clinical studies were pooled for a global efficacy and safety analysis. Baseline characteristics for the patients evaluated in the primary and secondary efficacy analyses were (mean, SD): chronological age (years): 6.7 ± 3.8; height (cm): 84.8 ± 15.3 cm; height standard deviation score (SDS): -6.7 ± 1.8; height velocity (cm/yr): 2.8 ± 1.8; height velocity SDS: -3.3 ± 1.7; IGF-1 (ng/mL): 21.6 ± 20.6; IGF-1 SDS: -4.3 ± 1.6; and bone age (years): 4.2 ± 2.8. Sixty-one subjects had at least one year of treatment. Fifty-three (87%) had Laron Syndrome; 7 (11%) had GH gene deletion, and 1 (2%) had neutralizing antibodies to GH. Thirty-seven (61%) of the subjects were male; forty-eight (79%) were Caucasian. Fifty-six (92%) of the subjects were pre-pubertal at baseline.
Annual results for height velocity, height velocity SDS, and height SDS are shown in Table 1. Pre-treatment height velocity data were available for 58 subjects. The height velocities at a given year of treatment were compared by paired t-tests to the pre-treatment height velocities of the same subjects completing that treatment year.
| Pre-Tx | Year 1 | Year 2 | Year 3 | Year 4 | Year 5 | Year 6 | Year 7 | Year 8 | |
| Pre-Tx = Pre-treatment; SD = Standard Deviation; SDS = Standard Deviation Score | |||||||||
| [1] P-values for comparison versus pre-Tx values are computed using paired t-tests. | |||||||||
| Height Velocity | |||||||||
| (cm/yr) | |||||||||
| N | 58 | 58 | 48 | 38 | 23 | 21 | 20 | 16 | 13 |
| Mean (SD) | 2.8 (1.8) | 8.0 (2.2) | 5.8 (1.5) | 5.5 (1.8) | 4.7 (1.6) | 4.7 (1.6) | 4.8 (1.5) | 4.6 (1.5) | 4.3 (1.1) |
| Mean (SD) for change | +5.2 | +2.9 | +2.3 | +1.5 | +1.5 | +1.5 | +1.0 | +0.7 | |
| from pre-Tx | (2.6) | (2.4) | (2.4) | (2.2) | (1.8) | (1.7) | (2.1) | (2.5) | |
| P-value for change | <0.0001 | <0.0001 | <0.0001 | 0.0045 | 0.0015 | 0.0009 | 0.0897 | 0.3059 | |
| from pre-Tx [1] | |||||||||
| Height Velocity SDS | |||||||||
| N | 58 | 58 | 47 | 37 | 22 | 19 | 18 | 15 | 11 |
| Mean (SD) | -3.3 (1.7) | 1.9 (3.0) | -0.2 | -0.2 | -0.7 | -0.6 | -0.4 | -0.4 | -0.4 |
| (1.6) | (2.0) | (2.1) | (2.1) | (1.4) | (1.9) | (1.9) | |||
| Mean (SD) for change | +5.2 | +3.1 | +2.9 | +2.2 | +2.5 | +2.7 | +2.5 | +2.7 | |
| from pre-Tx | (3.1) | (2.3) | (2.3) | (2.2) | (2.2) | (1.7) | (2.1) | (2.8) | |
| Height SDS | |||||||||
| N | 61 | 61 | 51 | 40 | 24 | 21 | 20 | 16 | 13 |
| Mean (SD) | -6.7 (1.8) | -5.9 | -5.6 | -5.4 | -5.5 | -5.6 | -5.4 | -5.2 | -5.2 |
| (1.8) | (1.8) | (1.8) | (1.9) | (1.8) | (1.8) | (2.0) | (2.0) | ||
| Mean (SD) for change | +0.8 | +1.2 | +1.4 | +1.3 | +1.4 | +1.4 | +1.4 | +1.5 | |
| from pre-Tx | (0.5) | (0.8) | (1.1) | (1.2) | (1.3) | (1.2) | (1.1) | (1.1) | |
Forty-nine subjects were included in an analysis of the effects of INCRELEX® on bone age advancement. The mean ± SD change in chronological age was 4.9 ± 3.4 years and the mean ± SD change in bone age was 5.3 ± 3.4 years.
INDICATIONS AND USAGE
INCRELEX® (mecasermin [rDNA origin] injection) is indicated for the long-term treatment of growth failure in children with severe primary IGF-1 deficiency (Primary IGFD) or with growth hormone (GH) gene deletion who have developed neutralizing antibodies to GH. Severe Primary IGFD is defined by:
-
height standard deviation score ≤ –3.0 and
-
basal IGF-1 standard deviation score ≤ –3.0 and
-
normal or elevated growth hormone (GH).
Severe Primary IGFD includes patients with mutations in the GH receptor (GHR), post-GHR signaling pathway, and IGF-1 gene defects; they are not GH deficient, and therefore, they cannot be expected to respond adequately to exogenous GH treatment.
INCRELEX® is not intended for use in subjects with secondary forms of IGF-1 deficiency, such as GH deficiency, malnutrition, hypothyroidism, or chronic treatment with pharmacologic doses of anti-inflammatory steroids. Thyroid and nutritional deficiencies should be corrected before initiating INCRELEX® treatment.
INCRELEX® is not a substitute for GH treatment.
CONTRAINDICATIONS
INCRELEX® should not be used for growth promotion in patients with closed epiphyses.
INCRELEX® is contraindicated in the presence of active or suspected neoplasia, and therapy should be discontinued if evidence of neoplasia develops.
Intravenous administration of INCRELEX® is contraindicated.
INCRELEX® should not be used by patients who are allergic to mecasermin (IGF-1) or any of the inactive ingredients in INCRELEX®.
WARNINGS
INCRELEX contains benzyl alcohol as a preservative. Benzyl alcohol as a preservative has been associated with neurologic toxicity in neonates.
If sensitivity to INCRELEX® occurs, treatment should be discontinued.
PRECAUTIONS
General
Treatment with INCRELEX® should be directed by physicians who are experienced in the diagnosis and management of patients with growth disorders.
Information for Patients
Patients and/or their parents should be instructed in the safe administration o f INCRELEX®. INCRELEX® should be given shortly before or after (20 minutes on either side of) a meal or snack. INCRELEX® should not be administered when the meal or snack is omitted. The dose of INCRELEX® should never be increased to make up for one or more omitted doses. INCRELEX® therapy should be initiated at a low dose and the dose should be increased only if no hypoglycemia episodes have occurred after at least 7 days of dosing. If severe hypoglycemia or persistent hypoglycemia occurs on treatment despite adequate food intake, INCRELEX® dose reduction should be considered. Providers should educate patients and caregivers on how to recognize the signs and symptoms of hypoglycemia.
Patients and/or parents should be thoroughly instructed in the importance of proper needle disposal. A puncture-resistant container should be used for the disposal of used needles and/or syringes (consistent with applicable state requirements). Needles and syringes must not be reused.
INCRELEX® has not been studied in children less than 2 years of age or in adults.
INCRELEX® should be administered shortly before or after a meal or snack, because it has insulin-like hypoglycemic effects. Special attention should be paid to small children because their oral intake may not be consistent. Patients should avoid engaging in any high-risk activities (e.g., driving, etc.) within 2-3 hours after dosing, particularly at the initiation of INCRELEX® treatment, until a well-tolerated dose of INCRELEX® has been established.
Lymphoid tissue (e.g., tonsillar) hypertrophy associated with complications, such as snoring, sleep apnea, and chronic middle-ear effusions have been reported with the use of INCRELEX®. Patients should have periodic examinations to rule out such potential complications and receive appropriate treatment if necessary.
Intracranial hypertension (IH) with papilledema, visual changes, headache, nausea and/or vomiting have been reported in patients treated with INCRELEX®, as they have been reported with therapeutic growth hormone administration. IH-associated signs and symptoms resolved after interruption of dosing. Funduscopic examination is recommended at the initiation and periodically during the course of INCRELEX® therapy.
Slipped capital femoral epiphysis and progression of scoliosis can occur in patients who experience rapid growth. These conditions and other symptoms and signs known to be associated with GH treatment in general should be monitored during INCRELEX® treatment.
As with any exogenous protein administration, local or systemic allergic reactions may occur. Parents and patients should be informed that such reactions are possible and that if an allergic reaction occurs, treatment should be interrupted and prompt medical attention should be sought.
Carcinogenesis, mutagenesis, impairment of fertility
INCRELEX® was administered subcutaneously to Sprague Dawley rats at doses of 0, 0.25, 1, 4, and 10 mg/kg/day for up to 2 years. An increased incidence of adrenal medullary hyperplasia and pheochromocytoma was observed in male rats at doses of 1 mg/kg/day and above (≥ 1 times the clinical exposure with the maximum recommended human dose [MRHD] based on AUC) and female rats at all dose levels (≥ 0.3 times the clinical exposure with the MRHD based on AUC). An increased incidence of keratoacanthoma in the skin was observed in male rats at doses of 4 and 10 mg/kg/day (≥ 4 times the MRHD) and in female rats treated with 10 mg/kg/day (7 times the MRHD based on AUC). An increased incidence of mammary gland carcinoma in both male and female rats was observed in animals treated with 10 mg/kg/day (7 times the MRHD based on AUC). Based on excess mortality secondary to IGF-1 induced hypoglycemia, these skin and mammary tumor findings were only observed at doses that exceeded the maximum tolerated dose (MTD).
Mutagenesis
INCRELEX® was not clastogenic in the in vitro chromosome aberration assay and the in vivo mouse micronucleus assay.
Impairment of fertility
INCRELEX® was administered intravenously to rats at doses of 0.25, 1, and 4 mg/day to conduct the fertility study. No effects on fertility were observed in male or female rats treated with doses up to 4 mg/kg/day (4 times the clinical exposure with the MRHD based on AUC.)
Pregnancy Category C.
Embryo-fetal toxicity studies were conducted in Sprague Dawley rats with doses of 1, 4, and 16 mg/kg/day, and in New Zealand White rabbits with doses of 0.125, 0.5, and 2 mg/kg/day administered intravenously. No embryo-fetal developmental abnormalities were observed in rats with doses up to 16 mg/kg/day (20 times the MRHD based on body surface area [BSA] comparison). In the rabbit study, the NOAEL for maternal toxicity was 2 mg/kg (8 times the MRHD based on BSA) and the NOAEL for fetal toxicity was 0.5 mg/kg (2 times the MRHD based on BSA). INCRELEX® displayed no teratogenicity at doses up to 2 mg/kg (8 times the MRHD based on BSA).
The effects of INCRELEX® on an unborn child have not been studied. Therefore, there is insufficient medical information to determine whether there are significant risks to a fetus.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when INCRELEX® is administered to a nursing woman.
Geriatric Use
The safety and effectiveness of INCRELEX® in patients aged 65 and over has not been evaluated in clinical studies.
ADVERSE REACTIONS
As with all protein pharmaceuticals, some patients may develop antibodies to INCRELEX®. Anti-IGF-1 antibodies were present at one or more of the periodic assessments in 14 of 23 children with Primary IGFD treated for 2 years. However, no clinical consequences of these antibodies were observed (e.g., allergic reactions or attenuation of growth).
In clinical studies of 71 subjects with Primary IGFD treated for a mean duration of 3.9 years and representing 274 subject-years, no subjects withdrew from any clinical study because of adverse events. Adverse events considered related to INCRELEX® treatment that occurred in 5% or more of these study participants are listed below by organ class.
Metabolism and Nutrition Disorders: hypoglycemia
General Disorders and Administrative Site Conditions: lipohypertrophy, bruising
Infections and Infestations: otitis media, serous otitis media
Respiratory, Thoracic and Mediastinal Disorders: snoring, tonsillar hypertrophy
Nervous System Disorders: headache, dizziness, convulsions
Gastrointestinal Disorders: vomiting
Ear and Labyrinth Disorders: hypoacusis, fluid in middle ear, ear pain, abnormal tympanometry
Cardiac Disorders: cardiac murmur
Musculoskeletal and Connective Tissue Disorders: arthralgia, pain in extremity
Blood and Lymphatic System Disorders: thymus hypertrophy
Surgical and Medical Procedures: ear tube insertion
Hypoglycemia was reported by 30 subjects (42%) at least once during their course of therapy. Most cases of hypoglycemia were mild or moderate in severity. Five subjects had severe hypoglycemia (requiring assistance and treatment) on one or more occasion and 4 subjects experienced hypoglycemic seizures/loss of consciousness on one or more occasion. Of the 30 subjects reporting hypoglycemia, 14 (47%) had a history of hypoglycemia prior to treatment. The frequency of hypoglycemia was highest in the first month of treatment, and episodes were more frequent in younger children. Symptomatic hypoglycemia was generally avoided when a meal or snack was consumed either shortly (i.e., 20 minutes) before or after the administration of INCRELEX®.
Tonsillar hypertrophy was noted in 11 (15%) subjects in the first 1 to 2 years of therapy with lesser tonsillar growth in subsequent years. Tonsillectomy or tonsillectomy/adenoidectomy was performed in 7 subjects; 3 of these had obstructive sleep apnea, which resolved after the procedure in all three cases.
Intracranial hypertension occurred in three subjects. In two subjects the events resolved without interruption of INCRELEX® treatment. INCRELEX® treatment was discontinued in the third subject and resumed later at a lower dose without recurrence.
Mild elevations in the serum AST and LDH were found in a significant proportion of patients before and during treatment and no rise in levels of these serum enzymes led to treatment discontinuation. ALT elevations were occasionally noted during treatment. Renal and splenic lengths (measured by ultrasound) increased rapidly on INCRELEX® treatment during the first years of therapy. This lengthening slowed down subsequently; though in some patients, renal and/or splenic length reached or surpassed the 95th percentile. Renal function (as defined by serum creatinine and calculated creatinine clearance) was normal in all patients, irrespective of renal growth. Elevations in cholesterol and triglycerides to above the upper limit of normal were observed before and during treatment. Echocardiographic evidence of cardiomegaly/valvulopathy was observed in a few individuals without associated clinical symptoms. Because of underlying disease and the lack of control group, the relation of the cardiac changes to drug treatment cannot be assessed.
Thickening of the soft tissues of the face was observed in several patients and should be monitored during INCRELEX® treatment.
OVERDOSAGE
There is no clinical experience with overdosage of INCRELEX®. Based on known pharmacological effects, acute overdosage would be predicted to lead to hypoglycemia. Long-term overdosage may result in signs and symptoms of acromegaly. Treatment of acute overdose of INCRELEX® should be directed at reversing hypoglycemia. Oral glucose or food should be consumed. If the overdose results in loss of consciousness, intravenous glucose or parenteral glucagon may be required to reverse the hypoglycemic effects.
DOSAGE AND ADMINISTRATION
Preprandial glucose monitoring should be considered at treatment initiation and until a well tolerated dose is established. If frequent symptoms of hypoglycemia or severe hypoglycemia occur, preprandial glucose monitoring should continue. The dosage of INCRELEX® should be individualized for each patient. The recommended starting dose of INCRELEX® is 0.04 to 0.08 mg/kg (40 to 80 μg/kg) twice daily by subcutaneous injection. If well-tolerated for at least one week, the dose may be increased by 0.04 mg/kg per dose, to the maximum dose of 0.12 mg/kg given twice daily. Doses greater than 0.12 mg/kg given twice daily have not been evaluated in children with Primary IGFD and, due to potential hypoglycemic effects, should not be used. If hypoglycemia occurs with recommended doses, despite adequate food intake, the dose should be reduced. INCRELEX® should be administered shortly before or after (± 20 minutes) a meal or snack. If the patient is unable to eat shortly before or after a dose for any reason, that dose of INCRELEX® should be withheld. Subsequent doses of INCRELEX® should never be increased to make up for one or more omitted dose.
INCRELEX® injection sites should be rotated to a different site with each injection.
INCRELEX® should be administered using sterile disposable syringes and needles. The syringes should be of small enough volume that the prescribed dose can be withdrawn from the vial with reasonable accuracy.
STABILITY AND STORAGE
Before Opening - Vials of INCRELEX® are stable when refrigerated [2º to 8ºC (35º to 46ºF)]. Avoid freezing the vials of INCRELEX®. Protect from direct light. Expiration dates are stated on the labels.
After Opening – Vials of INCRELEX® are stable for 30 days after initial vial entry when stored at 2º to 8ºC (35º to 46ºF). Avoid freezing the vials of INCRELEX®. Protect from direct light.
Vial contents should be clear without particulate matter. If the solution is cloudy or contains particulate matter, the contents must not be injected. INCRELEX® should not be used after its expiration date. Keep refrigerated and use within 30 days of initial vial entry. Remaining unused material should be discarded.
HOW SUPPLIED
INCRELEX® is supplied as a 10 mg/mL sterile solution in multiple dose glass vials (40 mg/vial).
NDC-15054-1040-5
Rx only
Manufactured for: Tercica, Inc.
Brisbane, CA 94005 USA
by: Hospira, Incorporated
McPherson, KS 67460 USA
Issued: August 2005
Revised: September 2008
TN5EN-00
PRINCIPAL DISPLAY PANEL - 40 mg/4mL Carton
NDC 15054-1040-5
One multi-use vial
Rx only
increlex®
(mecasermin [rDNA origin]
injection)
40 mg/4 mL
(10 mg/mL)
For subcutaneous use only
Net wt. 40 mg
| INCRELEX
mecasermin injection, solution |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021839 | 01/03/2006 | |
| Labeler - Tercica, Inc. (118461578) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Hospira, Inc. | 030606222 | MANUFACTURE | |