PERINDOPRIL ERBUMINE
-
perindopril erbumine tablet
Roxane Laboratories, Inc.
----------
Perindopril Erbumine Tablets, 2 mg, 4 mg and 8 mgRx only
|
USE IN PREGNANCY When used in pregnancy, ACE inhibitors can cause injury and even death to the developing fetus. When pregnancy is detected, Perindopril Erbumine Tablets should be discontinued as soon as possible. See WARNINGS: Fetal/Neonatal Morbidity and Mortality . |
DESCRIPTION
Perindopril Erbumine Tablets is the tert-butylamine salt of perindopril, the ethyl ester of a non-sulfhydryl angiotensin-converting enzyme (ACE) inhibitor. Perindopril erbumine is chemically described as (2S,3∝S,7∝S)-1-[(S)-N-[(S)-1-Carboxy-butyl]alanyl]hexahydro-2-indolinecarboxylic acid, 1-ethyl ester, compound with ter-butylamine (1:1). Its molecular formula is C19H32N2O5C4H11N. Its structural formula is:
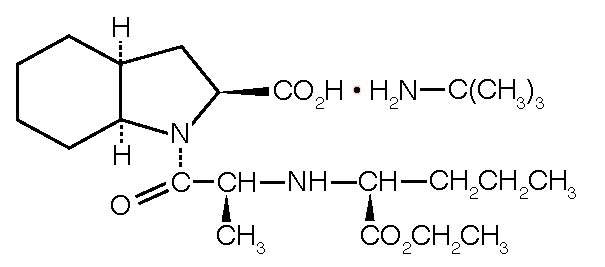
Perindopril erbumine is a white, crystalline powder with a molecular weight of 368.47 (free acid) or 441.61 (salt form). It is freely soluble in water (60% w/w), alcohol and chloroform.
Perindopril is the free acid form of perindopril erbumine, is a pro-drug and metabolized in vivo by hydrolysis of the ester group to form perindoprilat, the biologically active metabolite.
Perindopril Erbumine Tablets are available in 2 mg, 4 mg and 8 mg strengths for oral administration. In addition to perindopril erbumine, each tablet contains the following inactive ingredients: lactose (anhydrous), magnesium stearate, microcrystalline cellulose and silicon dioxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Perindopril erbumine tablets are a pro-drug for perindoprilat, which inhibits ACE in human subjects and animals. The mechanism through which perindoprilat lowers blood pressure is believed to be primarily inhibition of ACE activity. ACE is a peptidyl dipeptidase that catalyzes conversion of the inactive decapeptide, angiotensin I, to the vasoconstrictor, angiotensin II. Angiotensin II is a potent peripheral vasoconstrictor, which stimulates aldosterone secretion by the adrenal cortex, and provides negative feedback on renin secretion. Inhibition of ACE results in decreased plasma angiotensin II, leading to decreased vasoconstriction, increased plasma renin activity and decreased aldosterone secretion. The latter results in diuresis and natriuresis and may be associated with a small increase of serum potassium.
ACE is identical to kininase II, an enzyme that degrades bradykinin. Whether increased levels of bradykinin, a potent vasodepressor peptide, play a role in the therapeutic effects of perindopril erbumine tablets remains to be elucidated.
While the principal mechanism of perindopril in blood pressure reduction is believed to be through the renin-angiotensin-aldosterone system, ACE inhibitors have some effect even in apparent low-renin hypertension. Perindopril has been studied in relatively few black patients, usually a low-renin population, and the average response of diastolic blood pressure to perindopril was about half the response seen in nonblacks, a finding consistent with previous experience of other ACE inhibitors.
After administration of perindopril, ACE is inhibited in a dose and blood concentration-related fashion, with the maximal inhibition of 80 to 90% attained by 8 mg persisting for 10 to 12 hours. Twenty-four hour ACE inhibition is about 60% after these doses. The degree of ACE inhibition achieved by a given dose appears to diminish over time (the ID50 increases). The pressor response to an angiotensin I infusion is reduced by perindopril, but this effect is not as persistent as the effect on ACE; there is about 35% inhibition at 24 hours after a 12 mg dose.
Pharmacokinetics
Oral administration of perindopril erbumine tablets results in its rapid absorption with peak plasma concentrations occurring at approximately 1 hour. The absolute oral bioavailability of perindopril is about 75%. Following absorption, approximately 30 to 50% of systemically available perindopril is hydrolyzed to its active metabolite, perindoprilat, which has a mean bioavailability of about 25%. Peak plasma concentrations of perindoprilat are attained 3 to 7 hours after perindopril administration. The presence of food in the gastrointestinal tract does not affect the rate or extent of absorption of perindopril but reduces bioavailability of perindoprilat by about 35%. (See PRECAUTIONS: Food Interaction .)
With 4, 8 and 16 mg doses of perindopril erbumine tablets, Cmax and AUC of perindopril and perindoprilat increase in a linear and dose-proportional manner following both single oral dosing and at steady state during a once-a-day multiple dosing regimen.
Perindopril exhibits multiexponential pharmacokinetics following oral administration. The mean half-life of perindopril associated with most of its elimination is approximately 0.8 to 1 hour. At very low plasma concentrations of perindopril (<3 ng/mL), there is a prolonged terminal elimination half-life, similar to that seen with other ACE inhibitors, that results from slow dissociation of perindopril from plasma/tissue ACE binding sites. Perindopril does not accumulate with a once-a-day multiple dosing regimen. Mean total body clearance of perindopril is 219 to 362 mL/min and its mean renal clearance is 23.3 to 28.6 mL/min.
Perindopril is extensively metabolized following oral administration, with only 4 to 12% of the dose recovered unchanged in the urine. Six metabolites resulting from hydrolysis, glucuronidation and cyclization via dehydration have been identified. These include the active ACE inhibitor, perindoprilat (hydrolyzed perindopril), perindopril and perindoprilat glucuronides, dehydrated perindopril and the diastereoisomers of dehydrated perindoprilat. In humans, hepatic esterase appears to be responsible for the hydrolysis of perindopril.
The active metabolite, perindoprilat, also exhibits multiexponential pharmacokinetics following the oral administration of perindopril erbumine tablets. Formation of perindoprilat is gradual with peak plasma concentrations occurring between 3 and 7 hours. The subsequent decline in plasma concentration shows an apparent mean half-life of 3 to 10 hours for the majority of the elimination, with a prolonged terminal elimination half-life of 30 to 120 hours resulting from slow dissociation of perindoprilat from plasma/tissue ACE binding sites. During repeated oral once-daily dosing with perindopril, perindoprilat accumulates about 1.5 to 2 fold and attains steady state plasma levels in 3 to 6 days. The clearance of perindoprilat and its metabolites is almost exclusively renal.
Approximately 60% of circulating perindopril is bound to plasma proteins, and only 10 to 20% of perindoprilat is bound. Therefore, drug interactions mediated through effects on protein binding are not anticipated.
At usual antihypertensive dosages, little radioactivity (<5% of the dose) was distributed to the brain after administration of 14C-perindopril to rats.
Radioactivity was detectable in fetuses and in milk after administration of 14C-perindopril to pregnant and lactating rats.
Elderly Patients
Plasma concentrations of both perindopril and perindoprilat in elderly patients (>70 yrs) are approximately twice those observed in younger patients, reflecting both increased conversion of perindopril to perindoprilat and decreased renal excretion of perindoprilat. (See PRECAUTIONS: Geriatric Use.)
Heart Failure Patients
Perindoprilat clearance is reduced in congestive heart failure patients, resulting in a 40% higher dose interval AUC. (See DOSAGE AND ADMINISTRATION.)
Patients with Renal Insufficiency
With perindopril erbumine doses of 2 to 4 mg, perindoprilat AUC increases with decreasing renal function. At creatinine clearances of 30 to 80 mL/min, AUC is about double that of 100 mL/min. When creatinine clearance drops below 30 mL/min, AUC increases more markedly.
In a limited number of patients studied, perindopril dialysis clearance ranged from 41.7 to 76.7 mL/min (mean 52 mL/min). Perindoprilat dialysis clearance ranged from 37.4 to 91 mL/min (mean 67.2 mL/min). (See DOSAGE AND ADMINISTRATION.)
Patients with Hepatic Insufficiency
The bioavailability of perindoprilat is increased in patients with impaired hepatic function. Plasma concentrations of perindoprilat in patients with impaired liver function were about 50% higher than those observed in healthy subjects or hypertensive patients with normal liver function.
Pharmacodynamics and Clinical Effects
Stable Coronary Artery Disease
The EURopean trial On reduction of cardiac events with perindopril in stable coronary Artery disease (EUROPA) was a multicenter, randomized, double-blind and placebo-controlled study conducted in 12,218 patients who had evidence of stable coronary artery disease without clinical heart failure. Patients had evidence of coronary artery disease documented by previous myocardial infarction more than 3 months before screening, coronary revascularization more than 6 months before screening, angiographic evidence of stenosis (at least 70% narrowing of one or more major coronary arteries), or positive stress test in men with a history of chest pain. After a run-in period of 4 weeks during which all patients received perindopril 2 mg to 8 mg, the patients were randomly assigned to perindopril 8 mg once daily (N=6,110) or matching placebo (N=6,108). The mean follow-up was 4.2 years. The study examined the long-term effects of perindopril on time to first event of cardiovascular mortality, nonfatal myocardial infarction, or cardiac arrest in patients with stable coronary artery disease.
The mean age of patients was 60 years; 85% were male, 92% were taking platelet inhibitors, 63% were taking β blockers, and 56% were taking lipid-lowering therapy. The EUROPA study showed that perindopril significantly reduced the relative risk for the primary endpoint events (Table 1). This beneficial effect is largely attributable to a reduction in the risk of non-fatal myocardial infarction. This beneficial effect of perindopril on the primary outcome was evident after about one year of treatment (Figure 1).
| Perindopril (N=6,110) | Placebo (N=6,108) | RRR (95% CI) | P | |
| Combined Endpoint | ||||
| Cardiovascular Mortality, Non-Fatal MI, or Cardiac Arrest | 488 (8%) | 603 (9.9%) | 20% (9 to 29) | 0.0003 |
| Component Endpoint | ||||
| Cardiovascular Mortality | 215 (3.5%) | 249(4.1%) | 14% (-3 to 28) | 0.107 |
| Non-Fatal MI | 295 (4.8%) | 378 (6.2%) | 22% (10 to 33) | 0.001 |
| Cardiac Arrest | 6 (0.1%) | 11 (0.2%) | 46% (-47 to 80) | 0.22 |
RRR: relative risk reduction; MI: myocardial infarction
The outcome was similar across all predefined subgroups by age, underlying disease or concomitant medication (Figure 2).
Figure 1: Time to First Occurrence of Primary Endpoint
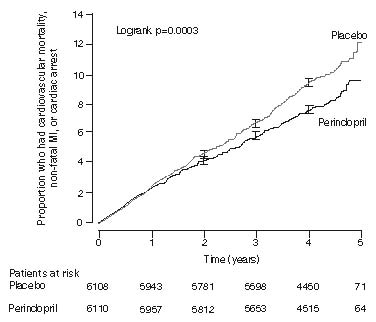
Figure 2: Beneficial Effect of Perindopril Treatment of Primary Endpoint in Predefined Subgroups
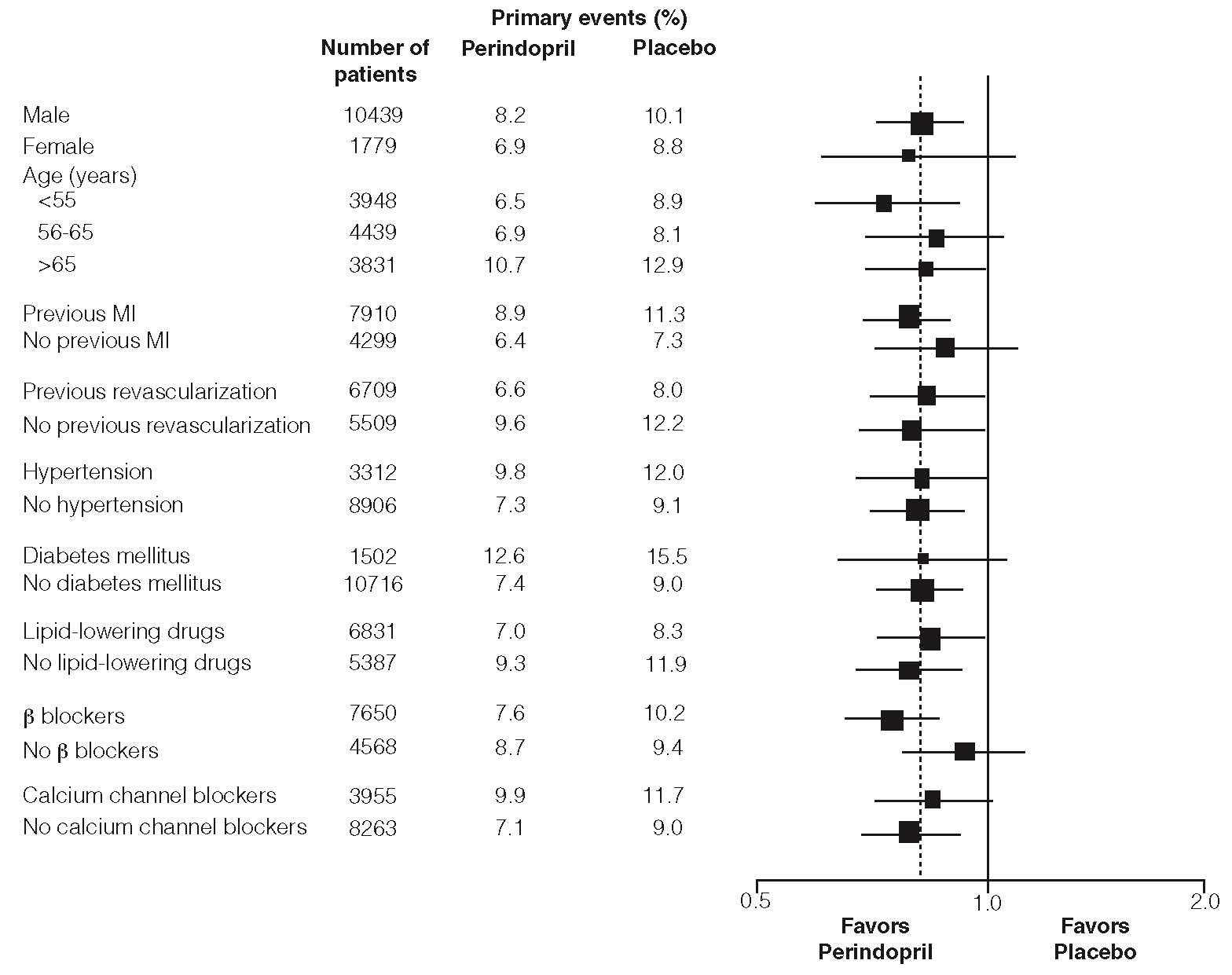
Size of squares proportional to the number of patients in that group. Dashed line indicates overall relative risk.
Hypertension
In placebo-controlled studies of perindopril monotherapy (2 to 16 mg q.d.) in patients with a mean blood pressure of about 150/100 mm Hg, 2 mg had little effect, but doses of 4 to 16 mg lowered blood pressure. The 8 and 16 mg doses were indistinguishable, and both had a greater effect than the 4 mg dose. The magnitude of the blood pressure effect was similar in the standing and supine positions, generally about 1 mm Hg greater on standing. In these studies, doses of 8 and 16 mg per day gave supine, trough blood pressure reductions of 9 to 15/5 to 6 mm Hg. When once-daily and twice-daily dosing were compared, the B.I.D. regimen was generally slightly superior, but by not more than about 0.5 to 1 mm Hg. After 2 to 16 mg doses of perindopril, the trough mean systolic and diastolic blood pressure effects were approximately equal to the peak effects (measured 3 to 7 hours after dosing). Trough effects were about 75 to 100% of peak effects. When perindopril was given to patients receiving 25 mg HCTZ, it had an added effect similar in magnitude to its effect as monotherapy, but 2 to 8 mg doses were approximately equal in effectiveness. In general, the effect of perindopril occurred promptly, with effects increasing slightly over several weeks.
In hemodynamic studies carried out in animal models of hypertension, blood pressure reduction after perindopril administration was accompanied by a reduction in peripheral arterial resistance and improved arterial wall compliance. In studies carried out in patients with essential hypertension, the reduction in blood pressure was accompanied by a reduction in peripheral resistance with no significant changes in heart rate or glomerular filtration rate. An increase in the compliance of large arteries was also observed, suggesting a direct effect on arterial smooth muscle, consistent with the results of animal studies.
Formal interaction studies of perindopril erbumine tablets have not been carried out with antihypertensive agents other than thiazides. Limited experience in controlled and uncontrolled trials coadministering perindopril erbumine tablets with a calcium channel blocker, a loop diuretic or triple therapy (beta-blocker, vasodilator and a diuretic), does not suggest any unexpected interactions. In general, ACE inhibitors have less than additive effects when given with beta-adrenergic blockers, presumably because both work in part through the renin angiotensin system. A controlled pharmacokinetic study has shown no effect on plasma digoxin concentrations when coadministered with perindopril erbumine tablets. (See PRECAUTIONS: Drug Interactions.)
In uncontrolled studies in patients with insulin-dependent diabetes, perindopril did not appear to affect glycemic control. In long-term use, no effect on urinary protein excretion was seen in these patients.
The effectiveness of perindopril erbumine tablets was not influenced by sex and it was less effective in blacks than in nonblacks. In elderly patients (≥60 years), the mean blood pressure effect was somewhat smaller than in younger patients, although the difference was not significant.
INDICATIONS AND USAGE
Stable Coronary Artery Disease
Perindopril Erbumine Tablets are indicated in patients with stable coronary artery disease to reduce the risk of cardiovascular mortality or non-fatal myocardial infarction. Perindopril Erbumine Tablets can be used with conventional treatment for management of coronary artery disease, such as antiplatelet, antihypertensive or lipid-lowering therapy.
Hypertension
Perindopril Erbumine Tablets are indicated for the treatment of patients with essential hypertension. Perindopril Erbumine Tablets may be used alone or given with other classes of antihypertensives, especially thiazide diuretics.
When using Perindopril Erbumine Tablets, consideration should be given to the fact that another angiotensin converting enzyme inhibitor (captopril) has caused agranulocytosis, particularly in patients with renal impairment or collagen vascular disease. Available data are insufficient to determine whether Perindopril Erbumine Tablets has a similar potential. (See WARNINGS.)
In considering use of Perindopril Erbumine Tablets, it should be noted that in controlled trials ACE inhibitors have an effect on blood pressure that is less in black patients than in nonblacks. In addition, it should be noted that black patients receiving ACE inhibitor monotherapy have been reported to have a higher incidence of angioedema compared to nonblacks. (See WARNINGS: Head and Neck Angioedema.)
CONTRAINDICATIONS
Perindopril Erbumine Tablets are contraindicated in patients known to be hypersensitive to this product or to any other ACE inhibitor. Perindopril Erbumine Tablets are also contraindicated in patients with a history of angioedema related to previous treatment with an ACE inhibitor.
WARNINGS
Anaphylactoid and Possibly Related Reactions
Presumably because angiotensin-converting enzyme inhibitors affect the metabolism of eicosanoids and polypeptides, including endogenous bradykinin, patients receiving ACE inhibitors (including perindopril erbumine tablets) may be subject to a variety of adverse reactions, some of them serious.
Head and Neck Angioedema
Angioedema involving the face, extremities, lips, tongue, glottis and/or larynx has been reported in patients treated with ACE inhibitors, including perindopril erbumine tablets (0.1% of patients treated with perindopril erbumine tablets in U.S. clinical trials). In such cases, perindopril erbumine tablets should be promptly discontinued and the patient carefully observed until the swelling disappears. In instances where swelling has been confined to the face and lips, the condition has generally resolved without treatment, although antihistamines have been useful in relieving symptoms. Angioedema associated with involvement of the tongue, glottis or larynx may be fatal due to airway obstruction. Appropriate therapy, such as subcutaneous epinephrine solution 1:1000 (0.3 to 0.5 mL), should be promptly administered. Patients with a history of angioedema unrelated to ACE inhibitor therapy may be at increased risk of angioedema while receiving an ACE inhibitor.
Intestinal Angioedema
Intestinal angioedema has been reported in patients treated with ACE inhibitors. These patients presented with abdominal pain (with or without nausea or vomiting); in some cases there was no prior history of facial angioedema and C-1 esterase levels were normal. The angioedema was diagnosed by procedures including abdominal CT scan or ultrasound, or at surgery, and symptoms resolved after stopping the ACE inhibitor. Intestinal angioedema should be included in the differential diagnosis of patients on ACE inhibitors presenting with abdominal pain.
Anaphylactoid Reactions During Desensitization
Two patients undergoing desensitizing treatment with hymenoptera venom while receiving ACE inhibitors sustained life-threatening anaphylactoid reactions. In the same patients, these reactions were avoided when ACE inhibitors were temporarily withheld, but they reappeared upon inadvertent rechallenge.
Anaphylactoid Reactions During Membrane Exposure
Anaphylactoid reactions have been reported in patients dialyzed with high-flux membranes and treated concomitantly with an ACE inhibitor. Anaphylactoid reactions have also been reported in patients undergoing low-density lipoprotein apheresis with dextran sulfate absorption.
Hypotension
Like other ACE inhibitors, perindopril erbumine tablets can cause symptomatic hypotension. Perindopril erbumine tablets has been associated with hypotension in 0.3% of uncomplicated hypertensive patients in U.S. placebo-controlled trials. Symptoms related to orthostatic hypotension were reported in another 0.8% of patients.
Symptomatic hypotension associated with the use of ACE inhibitors is more likely to occur in patients who have been volume and/or salt-depleted, as a result of prolonged diuretic therapy, dietary salt restriction, dialysis, diarrhea or vomiting. Volume and/or salt depletion should be corrected before initiating therapy with perindopril erbumine tablets. (See DOSAGE AND ADMINISTRATION.)
In patients with congestive heart failure, with or without associated renal insufficiency, ACE inhibitors may cause excessive hypotension, and may be associated with oliguria or azotemia, and rarely with acute renal failure and death. In patients with ischemic heart disease or cerebrovascular disease such an excessive fall in blood pressure could result in a myocardial infarction or a cerebrovascular accident.
In patients at risk of excessive hypotension, perindopril erbumine tablets therapy should be started under very close medical supervision. Patients should be followed closely for the first two weeks of treatment and whenever the dose of perindopril erbumine tablets and/or diuretic is increased.
If excessive hypotension occurs, the patient should be placed immediately in a supine position and, if necessary, treated with an intravenous infusion of physiological saline. Peridopril erbumine tablets treatment can usually be continued following restoration of volume and blood pressure.
Neutropenia/Agranulocytosis
Another ACE inhibitor, captopril, has been shown to cause agranulocytosis and bone marrow depression, rarely in uncomplicated patients but more frequently in patients with renal impairment, especially patients with a collagen vascular disease such as systemic lupus erythematosus or scleroderma. Available data from clinical trials of perindopril erbumine tablets are insufficient to show whether perindopril erbumine tablets causes agranulocytosis at similar rates.
Fetal/Neonatal Morbidity and Mortality
ACE inhibitors can cause fetal and neonatal morbidity and death when administered to pregnant women. Several dozen cases have been reported in the world literature. When pregnancy is detected, ACE inhibitors should be discontinued as soon as possible.
The use of ACE inhibitors during the second and third trimesters of pregnancy has been associated with fetal and neonatal injury, including hypotension, neonatal skull hypoplasia, anuria, reversible or irreversible renal failure and death. Oligohydramnios has also been reported, presumably resulting from decreased fetal renal function; oligohydramnios in this setting has been associated with fetal limb contractures, craniofacial deformation and hypoplastic lung development.
Prematurity, intrauterine growth retardation, patent ductus arteriosus, and other structural cardiac malformations, as well as neurological malformations, have been reported following exposure to ACE inhibitors during the first trimester of pregnancy.
When patients become pregnant, physicians should make every effort to discontinue the use of perindorpil erbumine tablets as soon as possible. Rarely (probably less often than once in every thousand pregnancies), no alternative to ACE inhibitors will be found. In these rare cases, the mothers should be apprised of the potential hazards to their fetuses, and serial ultrasound examinations should be performed to assess the intra-amniotic environment.
If oligohydramnios is observed, perindopril erbumine tablets should be discontinued unless it is considered life-saving for the mother. Contraction stress testing (CST), a non-stress test (NST) or biophysical profiling (BPP) may be appropriate, depending upon the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury.
Infants with histories of in utero exposure to ACE inhibitors should be closely observed for hypotension, oliguria and hyperkalemia. If oliguria occurs, attention should be directed toward support of blood pressure and renal perfusion. Exchange transfusion or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function. Perindopril, which crosses the placenta, can theoretically be removed from the neonatal circulation by these means, but limited experience has not shown that such removal is central to the treatment of these infants.
No teratogenic effects of perindopril were seen in studies of pregnant rats, mice, rabbits and cynomolgus monkeys. On a mg/m2 basis, the doses used in these studies were 6 times (in mice), 670 times (in rats), 50 times (in rabbits) and 17 times (in monkeys) the maximum recommended human dose (assuming a 50 kg adult). On a mg/kg basis, these multiples are 60 times (in mice), 3,750 times (in rats), 150 times (in rabbits) and 50 times (in monkeys) the maximum recommended human dose.
Hepatic Failure
Rarely, ACE inhibitors have been associated with a syndrome that starts with cholestatic jaundice and progresses to fulminant hepatic necrosis and (sometimes) death. The mechanism of this syndrome is not understood. Patients receiving ACE inhibitors who develop jaundice or marked elevations of hepatic enzymes should discontinue the ACE inhibitor and receive appropriate medical follow-up.
PRECAUTIONS
General
Impaired Renal Function
As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function may be anticipated in susceptible individuals.
Hypertensive Patients with Congestive Heart Failure
In patients with severe congestive heart failure, where renal function may depend on the activity of the renin-angiotensin-aldosterone system, treatment with ACE inhibitors, including perindopril erbumine tablets, may be associated with oliguria and/or progressive azotemia, and rarely with acute renal failure and/or death.
Hypertensive Patients with Renal Artery Stenosis
In hypertensive patients with unilateral or bilateral renal artery stenosis, increases in blood urea nitrogen and serum creatinine may occur. Experience with ACE inhibitors suggests that these increases are usually reversible upon discontinuation of the drug. In such patients, renal function should be monitored during the first few weeks of therapy.
Some hypertensive patients without apparent pre-existing renal vascular disease have developed increases in blood urea nitrogen and serum creatinine, usually minor and transient. These increases are more likely to occur in patients treated concomitantly with a diuretic and in patients with pre-existing renal impairment. Reduction of dosages of perindopril erbumine tablets, the diuretic or both may be required. In some cases, discontinuation of either or both drugs may be necessary.
Evaluation of hypertensive patients should always include an assessment of renal function. (See DOSAGE AND ADMINISTRATION.)
Hyperkalemia
Elevations of serum potassium have been observed in some patients treated with ACE inhibitors, including perindopril erbumine tablets. In U.S. controlled clinical trials, 1.4% of the patients receiving perindopril erbumine tablets and 2.3% of patients receiving placebo showed increased serum potassium levels to greater than 5.7 mEq/L. Most cases were isolated single values that did not appear clinically relevant and were rarely a cause for withdrawal. Risk factors for the development of hyperkalemia include renal insufficiency, diabetes mellitus and the concomitant use of agents such as potassium-sparing diuretics, potassium supplements and/or potassium-containing salt substitutes. Drugs associated with increases in serum potassium should be used cautiously, if at all, with perindopril erbumine tablets. (See PRECAUTIONS: Drug Interactions.)
Cough
Presumably due to the inhibition of the degradation of endogenous bradykinin, persistent nonproductive cough has been reported with all ACE inhibitors, always resolving after discontinuation of therapy. ACE inhibitor-induced cough should be considered in the differential diagnosis of cough. In controlled trials with perindopril, cough was present in 12% of perindopril patients and 4.5% of patients given placebo.
Surgery/Anesthesia
In patients undergoing surgery or during anesthesia with agents that produce hypotension, perindopril erbumine tablets may block angiotensin II formation that would otherwise occur secondary to compensatory renin release. Hypotension attributable to this mechanism can be corrected by volume expansion.
Information for Patients
Angioedema
Angioedema, including laryngeal edema, can occur with ACE inhibitor therapy, especially following the first dose. Patients should be told to report immediately signs or symptoms suggesting angioedema (swelling of face, extremities, eyes, lips, tongue, hoarseness or difficulty in swallowing or breathing) and to take no more drug before consulting a physician.
Symptomatic Hypotension
As with any antihypertensive therapy, patients should be cautioned that lightheadedness can occur, especially during the first few days of therapy and that it should be reported promptly. Patients should be told that if fainting occurs, perindopril erbumine tablets should be discontinued and a physician consulted.
All patients should be cautioned that inadequate fluid intake or excessive perspiration, diarrhea or vomiting can lead to an excessive fall in blood pressure in association with ACE inhibitor therapy.
Hyperkalemia
Patients should be advised not to use potassium supplements or salt substitutes containing potassium without a physician’s advice.
Neutropenia
Patients should be told to report promptly any indication of infection (e.g., sore throat, fever) which could be a sign of neutropenia.
Pregnancy
Female patients of childbearing age should be told about the consequences of exposure to ACE inhibitors during pregnancy. Discuss other treatment options with women planning to become pregnant. Women who do become pregnant while on an ACE inhibitor (including perindopril erbumine tablets) should be asked to stop the medication and contact their physician as soon as possible.
Drug Interactions
Diuretics
Patients on diuretics, and especially those started recently, may occasionally experience an excessive reduction of blood pressure after initiation of perindopril erbumine tablets therapy. The possibility of hypotensive effects can be minimized by either discontinuing the diuretic or increasing the salt intake prior to initiation of treatment with perindopril. If diuretics cannot be interrupted, close medical supervision should be provided with the first dose of perindopril erbumine tablets, for at least two hours and until blood pressure has stabilized for another hour. (See WARNINGS and DOSAGE AND ADMINISTRATION.)
The rate and extent of perindopril absorption and elimination are not affected by concomitant diuretics. The bioavailability of perindoprilat was reduced by diuretics, however, and this was associated with a decrease in plasma ACE inhibition.
Potassium Supplements and Potassium-Sparing Diuretics
Perindopril erbumine tablets may increase serum potassium because of its potential to decrease aldosterone production. Use of potassium-sparing diuretics (spironolactone, amiloride, triamterene and others), potassium supplements or other drugs capable of increasing serum potassium (indomethacin, heparin, cyclosporine and others) can increase the risk of hyperkalemia. Therefore, if concomitant use of such agents is indicated, they should be given with caution and the patient’s serum potassium should be monitored frequently.
Lithium
Increased serum lithium and symptoms of lithium toxicity have been reported in patients receiving concomitant lithium and ACE inhibitor therapy. These drugs should be coadministered with caution and frequent monitoring of serum lithium concentration is recommended. Use of a diuretic may further increase the risk of lithium toxicity.
Gold
Nitritoid reactions (symptoms include facial flushing, nausea, vomiting, and hypertension) have been reported rarely in patients on therapy with injectable gold (sodium aurothiomate) and concomitant ACE inhibitor therapy including perindopril erbumine tablets.
Digoxin
A controlled pharmacokinetic study has shown no effect on plasma digoxin concentrations when coadministered with perindopril erbumine tablets, but an effect of digoxin on the plasma concentration of perindopril/perindoprilat has not been excluded.
Gentamicin
Animal data have suggested the possibility of interaction between perindopril and gentamicin. However, this has not been investigated in human studies. Coadministration of both drugs should proceed with caution.
Food Interaction
Oral administration of perindopril erbumine tablets with food does not significantly lower the rate or extent of perindopril absorption relative to the fasted state. However, the extent of biotransformation of perindopril to the active metabolite, perindoprilat, is reduced approximately 43%, resulting in a reduction in the plasma ACE inhibition curve of approximately 20%, probably clinically insignificant. In clinical trials, perindopril was generally administered in a non-fasting state.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No evidence of carcinogenic effect was observed in studies in rats and mice when perindopril was administered at dosages up to 20 times (mg/kg) or 2 to 4 times (mg/m2) the maximum proposed clinical doses (16 mg/day) for 104 weeks.
Mutagenesis
No genotoxic potential was detected for perindopril erbumine tablets, perindoprilat and other metabolites in various in vitro and in vivo investigations, including the Ames test, the Saccharomycescerevisiae D4 test, cultured human lymphocytes, TK ± mouse lymphoma assay, mouse and rat micronucleus tests and Chinese hamster bone marrow assay.
Impairment of Fertility
There was no meaningful effect on reproductive performance or fertility in the rat given up to 30 times (mg/kg) or 6 times (mg/m2) the proposed maximum clinical dosage of perindopril erbumine tablets during the period of spermatogenesis in males or oogenesis and gestation in females.
Pregnancy
Pregnancy Category D. (See WARNINGS: Fetal/Neonatal Morbidity and Mortality .)
Nursing Mothers
Milk of lactating rats contained radioactivity following administration 14C-perindopril. It is not known whether perindopril is secreted in human milk. Because many drugs are secreted in human milk, caution should be exercised when perindopril erbumine tablets is given to nursing mothers.
Pediatric Use
Safety and effectiveness of perindopril erbumine tablets in pediatric patients have not been established.
Geriatric Use
The mean blood pressure effect of perindopril was somewhat smaller in patients over 60 than in younger patients, although the difference was not significant. Plasma concentrations of both perindopril and perindoprilat were increased in elderly patients compared to concentrations in younger patients. No adverse effects were clearly increased in older patients with the exception of dizziness and possibly rash.
Perindopril should be used with caution when administered to elderly patients who are at an increased risk for falls due to age, their underlying disease and/or their concomitant use of medication(s) associated with falls. Falls and fall-related events may be exacerbated by the central nervous system effects of dizziness and syncope as well as the symptomatic hypotension, including orthostatic, associated with perindopril. Experience with perindopril erbumine tablets in elderly patients at daily doses exceeding 8 mg is limited.
ADVERSE REACTIONS
Hypertension
Perindopril erbumine tablets has been evaluated for safety in approximately 3,400 patients with hypertension in U.S. and foreign clinical trials. Perindopril erbumine tablets was in general well-tolerated in the patient populations studied, the side effects were usually mild and transient. Although dizziness was reported more frequently in placebo patients (8.5%) than in perindopril patients (8.2%), the incidence appeared to increase with an increase in perindopril dose.
The data presented here are based on results from the 1,417 perindopril erbumine tablets-treated patients who participated in the U.S. clinical trials. Over 220 of these patients were treated with perindopril erbumine tablets for at least one year.
In placebo-controlled U.S. clinical trials, the incidence of premature discontinuation of therapy due to adverse events was 6.5% in patients treated with perindopril erbumine tablets and 6.7% in patients treated with placebo. The most common causes were cough, headache, asthenia and dizziness.
Among 1,012 patients in placebo-controlled U.S. trials, the overall frequency of reported adverse events was similar in patients treated with perindopril erbumine tablets and in those treated with placebo (approximately 75% in each group). Adverse events that occurred in 1% or greater of the patients and that were more common for perindopril than placebo by at least 1% (regardless of whether they were felt to be related to study drug) are shown in the first two columns below. Of these adverse events, those considered possibly or probably related to study drug are shown in the last two columns.
| All Adverse Events | Possibly-or Probably-Related Adverse Events | |||
| Perindopril (N=789) | Placebo (N=223) | Perindopril (N=789) | Placebo (N=223) | |
| Cough | 12 | 4.5 | 6 | 1.8 |
| Back Pain | 5.8 | 3.1 | 0 | 0 |
| Sinusitis | 5.2 | 3.6 | 0.6 | 0 |
| Viral Infection | 3.4 | 1.6 | 0.3 | 0 |
| Upper Extremity Pain | 2.8 | 1.4 | 0.2 | 0 |
| Hypertonia | 2.7 | 1.4 | 0.2 | 0 |
| Dyspepsia | 1.9 | 0.9 | 0.3 | 0 |
| Fever | 1.5 | 0.5 | 0.3 | 0 |
| Proteinuria | 1.5 | 0.5 | 1 | 0.5 |
| Ear Infection | 1.3 | 0 | 0 | 0 |
| Palpitation | 1.1 | 0 | 0.9 | 0 |
Of these, cough was the reason for withdrawal in 1.3% of perindopril and 0.4% of placebo patients. While dizziness was not reported more frequently in the perindopril group (8.2%) than in the placebo group (8.5%), it was clearly increased with dose, suggesting a causal relationship with perindopril. Other commonly reported complaints (1% or greater), regardless of causality, include: headache (23.8%), upper respiratory infection (8.6%), asthenia (7.9%), rhinitis (4.8%), low extremity pain (4.7%), diarrhea (4.3%), edema (3.9%), pharyngitis (3.3%), urinary tract infection (2.8%), abdominal pain (2.7%), sleep disorder (2.5%), chest pain (2.4%), injury, paresthesia, nausea, rash (each 2.3%), seasonal allergy, depression (each 2%), abnormal ECG (1.8%), ALT increase (1.7%), tinnitus, vomiting (each 1.5%), neck pain, male sexual dysfunction (each 1.4%), triglyceride increase, somnolence (each 1.3%), joint pain, nervousness, myalgia, menstrual disorder (each 1.1%), flatulence and arthritis (each 1%), but none of those was more frequent by at least 1% on perindopril than on placebo. Depending on the specific adverse event, approximately 30 to 70% of the common complaints were considered possibly or probably related to treatment.
Stable Coronary Artery Disease
Perindopril has been evaluated for safety in EUROPA, a double-blind, placebo-controlled study in 12,218 patients with stable coronary artery disease. The overall rate of discontinuation was about 22% on drug and placebo. The most common medical reasons for discontinuation that were more frequent on perindopril than placebo were cough, drug intolerance and hypotension.
Below is a list (by body system) of adverse experiences reported in 0.3 to 1% of patients in U.S. placebo-controlled studies in hypertensive patients without regard to attribution to therapy. Less frequent but medically important adverse events are also included; the incidence of these events is given in parentheses.
Body as a Whole: malaise, pain, cold/hot sensation, chills, fluid retention, orthostatic symptoms, anaphylactic reaction, facial edema, angioedema (0.1%).
Gastrointestinal: constipation, dry mouth, dry mucous membrane, appetite increased, gastroenteritis.
Respiratory: posterior nasal drip, bronchitis, rhinorrhea, throat disorder, dyspnea, sneezing, epistaxis, hoarseness, pulmonary fibrosis (<0.1%).
Urogenital: vaginitis, kidney stone, flank pain, urinary frequency, urinary retention.
Cardiovascular: hypotension, ventricular extrasystole, myocardial infarction, vasodilation, syncope, abnormal conduction, heart murmur, orthostatic hypotension.
Endocrine: gout.
Hematology: hematoma, ecchymosis.
Musculoskeletal: arthralgia, myalgia.
CNS: migraine, amnesia, vertigo, cerebral vascular accident (0.2%).
Psychiatric: anxiety, psychosexual disorder.
Dermatology: sweating, skin infection, tinea, pruritus, dry skin, erythema, fever blisters, purpura (0.1%).
Special Senses: conjunctivitis, earache.
Laboratory: potassium decrease, uric acid increase, alkaline phosphatase increase, cholesterol increase, AST increase, creatinine increase, hematuria, glucose increase.
When perindopril erbumine tablets were given concomitantly with thiazide diuretics, adverse events were generally reported at the same rate as those for perindopril erbumine tablets alone, except for a higher incidence of abnormal laboratory findings known to be related to treatment with thiazide diuretics alone (e.g., increases in serum uric acid, triglycerides and cholesterol and decreases in serum potassium).
Potential Adverse Effects Reported with ACE Inhibitors
Other medically important adverse effects reported with other available ACE inhibitors include: cardiac arrest, eosinophilic pneumonitis, neutropenia/agranulocytosis, pancytopenia, anemia (including hemolytic and aplastic), thrombocytopenia, acute renal failure, nephritis, hepatic failure, jaundice (hepatocellular or cholestatic), symptomatic hyponatremia, bullous pemphigoid, pemphigus, acute pancreatitis, falls, psoriasis, exfoliative dermatitis and a syndrome which may include: arthralgia/arthritis, vasculitis, serositis, myalgia, fever, rash or other dermatologic manifestations, a positive ANA, leukocytosis, eosinophilia or an elevated ESR. Many of these adverse effects have also been reported for perindopril.
Fetal/Neonatal Morbidity and Mortality
See WARNINGS: Fetal/Neonatal Morbidity and Mortality .
Clinical Laboratory Test Findings
Hypertension
Hematology, clinical chemistry and urinalysis parameters have been evaluated in U.S. placebo-controlled trials. In general, there were no clinically significant trends in laboratory test findings.
Hyperkalemia
In clinical trials, 1.4% of the patients receiving perindopril erbumine tablets and 2.3% of the patients receiving placebo showed serum potassium levels greater than 5.7 mEq/L. (See PRECAUTIONS.)
BUN/Serum Creatinine Elevations
Elevations, usually transient and minor, of BUN and serum creatinine have been observed. In placebo-controlled clinical trials, the proportion of patients experiencing increases in serum creatinine were similar in the perindopril erbumine tablets and placebo treatment groups. Rapid reduction of long-standing or markedly elevated blood pressure by any antihypertensive therapy can result in decreases in the glomerular filtration rate and, in turn, lead to increases in BUN or serum creatinine. (See PRECAUTIONS.)
Hematology
Small decreases in hemoglobin and hematocrit occur frequently in hypertensive patients treated with perindopril erbumine tablets, but are rarely of clinical importance. In controlled clinical trials, no patient was discontinued from therapy due to the development of anemia. Leukopenia (including neutropenia) was observed in 0.1% of patients in U.S. clinical trials (See WARNINGS.)
Liver Function Tests
Elevations in ALT (1.6% perindopril erbumine tablets vs 0.9% placebo) and AST (0.5% perindopril erbumine tablets vs 0.4% placebo) have been observed in U.S. placebo-controlled clinical trials. The elevations were generally mild and transient and resolved after discontinuation of therapy.
OVERDOSAGE
In animals, doses of perindopril up to 2,500 mg/kg in mice, 3,000 mg/kg in rats and 1,600 mg/kg in dogs were non-lethal. Past experiences were scant but suggested that overdosage with other ACE inhibitors was also fairly well tolerated by humans. The most likely manifestation is hypotension, and treatment should be symptomatic and supportive. Therapy with the ACE inhibitor should be discontinued, and the patient should be observed. Dehydration, electrolyte imbalance and hypotension should be treated by established procedures.
However, of the reported cases of perindopril overdosage, one (dosage unknown) required assisted ventilation and the other developed hypothermia, circulatory arrest and died following ingestion of up to 180 mg of perindopril. The intervention for perindopril overdose may require vigorous support (see below).
Laboratory determinations of serum levels of perindopril and its metabolites are not widely available, and such determinations have, in any event, no established role in the management of perindopril overdose.
No data are available to suggest physiological maneuvers (e.g., maneuvers to change the pH of the urine) that might accelerate elimination of perindopril and its metabolites. Perindopril can be removed by hemodialysis, with clearance of 52 mL/min for perindopril and 67 mL/min for perindoprilat.
Angiotensin II could presumably serve as a specific antagonist-antidote in the settling of perindopril overdose, but angiotensin II is essentially unavailable outside of scattered research facilities. Because the hypotensive effect of perindopril is achieved through vasodilation and effective hypovolemia, it is reasonable to treat perindopril overdose by infusion of normal saline solution.
DOSAGE AND ADMINISTRATION
Stable Coronary Artery Disease
In patients with stable coronary artery disease, perindopril erbumine tablets should be given at an initial dose of 4 mg once daily for 2 weeks, and then increased as tolerated, to a maintenance dose of 8 mg once daily. In elderly patients (>70 yrs), perindopril erbumine tablets should be given as a 2 mg dose once daily in the first week, followed by 4 mg once daily in the second week and 8 mg once daily for maintenance dose if tolerated.
Hypertension
Use in Uncomplicated Hypertensive Patients
In patients with essential hypertension, the recommended initial dose is 4 mg once a day. The dosage may be titrated upward until blood pressure, when measured just before the next dose, is controlled or to a maximum of 16 mg per day. The usual maintenance dose range is 4 to 8 mg administered as a single daily dose. Perindopril erbumine tablets may also be administered in two divided doses. When once-daily dosing was compared to twice-daily dosing in clinical studies, the B.I.D. regimen was generally slightly superior, but not by more than about 0.5 to 1 mm Hg.
Use in the Elderly Patients
As in younger patients, the recommended initial daily dosage of perindopril erbumine tablets for the elderly (>65 years) is 4 mg daily, given in one or two divided doses. The daily dosage may be titrated upward until blood pressure, when measured just before the next dose, is controlled, but experience with perindopril erbumine tablets is limited in the elderly at doses exceeding 8 mg. Dosages above 8 mg should be administered with caution and under close medical supervision. (See PRECAUTIONS: Geriatric Use.)
Use in Concomitant Diuretics
If blood pressure is not adequately controlled with perindopril alone, a diuretic may be added. In patients currently being treated with a diuretic, symptomatic hypotension occasionally can occur following the initial dose of perindopril. To reduce likelihood of such reaction, the diuretic should, if possible, be discontinued 2 to 3 days prior to the beginning of perindopril erbumine tablets therapy. (See WARNINGS.) Then, if blood pressure is not controlled with perindopril erbumine tablets alone, the diuretic should be resumed.
If the diuretic cannot be discontinued, an initial dose of 2 to 4 mg daily in one or in two divided doses should be used with careful medical supervision for several hours and until blood pressure has stabilized. The dosage should then be titrated as described above. (See WARNINGS and PRECAUTIONS: Drug Interactions.)
After the first dose of perindopril erbumine tablets, the patient should be followed closely for the first two weeks of treatment and whenever the dose of perindopril erbumine tablets and/or diuretics is increased (See WARNINGS and PRECAUTIONS: Drug Interactions.) In patients who are currently being treated with a diuretic, symptomatic hypotension occasionally can occur following the initial dose of perindopril erbumine tablets. To reduce the likelihood of hypotension, the dose of diuretic, if possible, can be adjusted which may diminish the likelihood of hypotension. The appearance of hypotension after the initial dose of perindopril erbumine tablets does not preclude subsequent careful dose titration with the drug, following effective management of the hypotension.
Dose Adjustment in Renal Impairment
Kinetic data indicate that perindoprilat elimination is decreased in renally impaired patients, with a marked increase in accumulation when creatinine clearance drops below 30 mL/min. In such patients (creatinine clearance <30 mL/min), safety and efficacy of perindopril erbumine tablets have not been established. For patients with lesser degrees of impairment (creatinine clearance above 30 mL/min), the initial dosage should be 2 mg/day and dosage should not exceed 8 mg/day due to limited clinical experience. During dialysis, perindopril is removed with the same clearance as in patients with normal renal function.
HOW SUPPLIED
Perindopril Erbumine Tablets are supplied as white to off-white, round, biconvex tablets. The 2 mg tablets are debossed with product identification "54 551" on one side and scored on the other side. The 4 mg tablets are debossed with product identification "54 327" on one side and scored on the other side. The 8 mg tablets are debossed with product identification "54 715" on one side and scored on the other side.
| 0054-0110-13 | 2 mg, white to off-white tablet, bottle of 30 |
| 0054-0110-25 | 2 mg, white to off-white tablet, bottle of 100 |
| 0054-0111-13 | 4 mg, white to off-white tablet, bottle of 30 |
| 0054-0111-25 | 4 mg, white to off-white tablet, bottle of 100 |
| 0054-0111-29 | 4 mg, white to off-white tablet, bottle of 500 |
| 0054-0112-13 | 8 mg, white to off-white tablet, bottle of 30 |
| 0054-0112-25 | 8 mg, white to off-white tablet, bottle of 100 |
| 0054-0112-29 | 8 mg, white to off-white tablet, bottle of 500 |
Storage
Store at controlled room temperature 20° to 25°C (68° to 77°F). [See USP Controlled Room Temperature.] Protect from moisture. Keep out of the reach of children.
10005475/01 Revised May 2009
© RLI, 2009
Package Label - Perindopril Erbumine Tablets
0054-0110-25
2 mg tablet, bottle of 100
Rx Only
Roxane Laboratories, Inc.
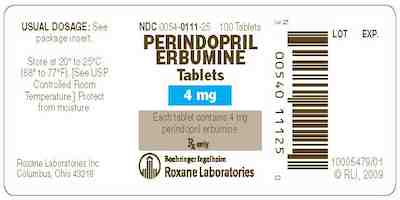
0054-0111-25
4 mg tablet, bottle of 100
Rx Only
Roxane Laboratories, Inc.
0054-0112-25
8 mg tablet, bottle of 100
Rx Only
Roxane Laboratories, Inc.

| PERINDOPRIL ERBUMINE
perindopril erbumine tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| ANDA | ANDA090072 | 11/10/2009 | |
| PERINDOPRIL ERBUMINE
perindopril erbumine tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| ANDA | ANDA090072 | 11/10/2009 | |
| PERINDOPRIL ERBUMINE
perindopril erbumine tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| ANDA | ANDA090072 | 11/10/2009 | |
| Labeler - Roxane Laboratories, Inc. (058839929) |
| Registrant - Roxane Laboratories, Inc. (058839929) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Boehringer Ingelheim Roxane Inc | 128407710 | MANUFACTURE | |