HYCAMTIN
-
topotecan hydrochloride injection, powder, lyophilized, for solution
SmithKline Beecham Corporation
----------
HYCAMTIN®(topotecan hydrochloride)
For Injection
FOR INTRAVENOUS USE
WARNING
HYCAMTIN (topotecan hydrochloride) for Injection should be administered under the supervision of a physician experienced in the use of cancer chemotherapeutic agents. Appropriate management of complications is possible only when adequate diagnostic and treatment facilities are readily available.
Therapy with HYCAMTIN should not be given to patients with baseline neutrophil counts of less than 1,500 cells/mm3. In order to monitor the occurrence of bone marrow suppression, primarily neutropenia, which may be severe and result in infection and death, frequent peripheral blood cell counts should be performed on all patients receiving HYCAMTIN.
DESCRIPTION
HYCAMTIN (topotecan hydrochloride) is a semi-synthetic derivative of camptothecin and is an anti-tumor drug with topoisomerase I-inhibitory activity.
HYCAMTIN for Injection is supplied as a sterile lyophilized, buffered, light yellow to greenishpowder available in single-dose vials. Each vial contains topotecan hydrochloride equivalent to 4 mg of topotecan as free base. The reconstituted solution ranges in color from yellow to yellow-green and is intended for administration by intravenous infusion.
Inactive ingredients are mannitol, 48 mg, and tartaric acid, 20 mg. Hydrochloric acid and sodium hydroxide may be used to adjust the pH. The solution pH ranges from 2.5 to 3.5.
The chemical name for topotecan hydrochloride is (S)-10-[(dimethylamino)methyl]-4-ethyl-4,9-dihydroxy-1H-pyrano[3’,4’:6,7] indolizino [1,2-b]quinoline-3,14-(4H ,12H)-dione monohydrochloride. It has the molecular formula C23H23N3O5•HCl and a molecular weight of 457.9.
Topotecan hydrochloride has the following structural formula:
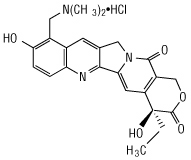
It is soluble in water and melts with decomposition at 213° to 218°C.
CLINICAL PHARMACOLOGY
Mechanism of Action
Topoisomerase I relieves torsional strain in DNA by inducing reversible single strand breaks. Topotecan binds to the topoisomerase I-DNA complex and prevents religation of these single strand breaks. The cytotoxicity of topotecan is thought to be due to double strand DNA damage produced during DNA synthesis, when replication enzymes interact with the ternary complex formed by topotecan, topoisomerase I, and DNA. Mammalian cells cannot efficiently repair these double strand breaks.
Pharmacokinetics
The pharmacokinetics of topotecan have been evaluated in cancer patients following doses of 0.5 to 1.5 mg/m2 administered as a 30-minute infusion. Topotecan exhibits multiexponential pharmacokinetics with a terminal half-life of 2 to 3 hours. Total exposure (AUC) is approximately dose-proportional. Binding of topotecan to plasma proteins is about 35%.
Metabolism and Elimination
Topotecan undergoes a reversible pH dependent hydrolysis of its lactone moiety; it is the lactone form that is pharmacologically active. At pH ≤4, the lactone is exclusively present, whereas the ring-opened hydroxy-acid form predominates at physiologic pH. In vitro studies in human liver microsomes indicate topotecan is metabolized to an N-demethylated metabolite. The mean metabolite:parent AUC ratio was about 3% for total topotecan and topotecan lactone following IV administration.
Renal clearance is an important determinant of topotecan elimination (see Special Populations: Renal Impairment).
In a mass balance/excretion study in 4 patients with solid tumors, the overall recovery of total topotecan and its N-desmethyl metabolite in urine and feces over 9 days averaged 73.4 ± 2.3% of the administered IV dose. Mean values of 50.8 ± 2.9% as total topotecan and 3.1 ± 1.0% as N-desmethyl topotecan were excreted in the urine following IV administration. Fecal elimination of total topotecan accounted for 17.9 ± 3.6% while fecal elimination of N-desmethyl topotecan was 1.7 ± 0.6%. An O-glucuronidation metabolite of topotecan and N-desmethyl topotecan has been identified in the urine. These metabolites, topotecan-O-glucuronide and N-desmethyl topotecan-O-glucuronide, were less than 2% of the administered dose.
Special Populations
Gender
The overall mean topotecan plasma clearance in male patients was approximately 24% higher than that in female patients, largely reflecting difference in body size.
Geriatrics
Topotecan pharmacokinetics have not been specifically studied in an elderly population, but population pharmacokinetic analysis in female patients did not identify age as a significant factor. Decreased renal clearance, which is common in the elderly, is a more important determinant of topotecan clearance (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Race
The effect of race on topotecan pharmacokinetics has not been studied.
Renal Impairment
In patients with mild renal impairment (creatinine clearance of 40 to 60 mL/min.), topotecan plasma clearance was decreased to about 67% of the value in patients with normal renal function. In patients with moderate renal impairment (Clcr of 20 to 39 mL/min.), topotecan plasma clearance was reduced to about 34% of the value in control patients, with an increase in half-life. Mean half-life, estimated in 3 renally impaired patients, was about 5.0 hours. Dosage adjustment is recommended for these patients (see DOSAGE AND ADMINISTRATION).
Hepatic Impairment
Plasma clearance in patients with hepatic impairment (serum bilirubin levels between 1.7 and 15.0 mg/dL) was decreased to about 67% of the value in patients without hepatic impairment. Topotecan half-life increased slightly, from 2.0 hours to 2.5 hours, but these hepatically impaired patients tolerated the usual recommended topotecan dosage regimen (see DOSAGE AND ADMINISTRATION).
Drug Interactions
Pharmacokinetic studies of the interaction of topotecan with concomitantly administered medications have not been formally investigated. In vitro inhibition studies using marker substrates known to be metabolized by human P450 CYP1A2, CYP2A6, CYP2C8/9, CYP2C19, CYP2D6, CYP2E, CYP3A, or CYP4A or dihydropyrimidine dehydrogenase indicate that the activities of these enzymes were not altered by topotecan. Enzyme inhibition by topotecan has not been evaluated in vivo.
Administration of cisplatin (60 or 75 mg/m2 on day 1) before topotecan (0.75 mg/m2/day on days 1-5) in 9 patients with ovarian cancer had no significant effect on the Cmax and AUC of total topotecan.
Topotecan had no effect on the pharmacokinetics of free platinum in 15 patients with ovarian cancer who were administered cisplatin 50 mg/m2 (n = 9) or 75 mg/m2 (n = 6) on day 2 after paclitaxel 110 mg/m2 on day 1 before topotecan 0.3 mg/m2 IV daily on days 2-6. Topotecan had no effect on dose-normalized (60 mg/m2) Cmax values of free platinum in 13 patients with ovarian cancer who were administered 60 mg/m2 (n = 10) or 75 mg/m2 (n = 3) cisplatin on day 1 before topotecan 0.75 mg/m2 IV daily on days 1-5.
No pharmacokinetic data are available following topotecan (0.75 mg/m2/day for 3 consecutive days) and cisplatin (50 mg/m2/day on day 1) in patients with cervical cancer.
Pharmacodynamics
The dose-limiting toxicity of topotecan is leukopenia. White blood cell count decreases with increasing topotecan dose or topotecan AUC. When topotecan is administered at a dose of 1.5 mg/m2/day for 5 days, an 80% to 90% decrease in white blood cell count at nadir is typically observed after the first cycle of therapy.
CLINICAL STUDIES
Ovarian Cancer
HYCAMTIN was studied in 2 clinical trials of 223 patients given topotecan with metastatic ovarian carcinoma. All patients had disease that had recurred on, or was unresponsive to, a platinum-containing regimen. Patients in these 2 studies received an initial dose of 1.5 mg/m2 given by intravenous infusion over 30 minutes for 5 consecutive days, starting on day 1 of a 21-day course.
One study was a randomized trial of 112 patients treated with HYCAMTIN (1.5 mg/m2/day × 5 days starting on day 1 of a 21-day course) and 114 patients treated with paclitaxel (175 mg/m2 over 3 hours on day 1 of a 21-day course). All patients had recurrent ovarian cancer after a platinum-containing regimen or had not responded to at least 1 prior platinum-containing regimen. Patients who did not respond to the study therapy, or who progressed, could be given the alternative treatment.
Response rates, response duration, and time to progression are shown in Table 1.
| Parameter |
HYCAMTIN (n = 112) |
Paclitaxel (n = 114) |
| Complete response rate | 5% | 3% |
| Partial response rate | 16% | 11% |
| Overall response rate | 21% | 14% |
| 95% Confidence interval | 13 to 28% | 8 to 20% |
| (p-value) | (0.20) | |
| Response duration* (weeks) | n = 23 | n = 16 |
| Median | 25.9 | 21.6 |
| 95% Confidence interval | 22.1 to 32.9 | 16.0 to 34.0 |
| hazard-ratio | ||
| (HYCAMTIN:paclitaxel) | 0.78 | |
| (p-value) | (0.48) | |
| Time to progression (weeks) | ||
| Median | 18.9 | 14.7 |
| 95% Confidence interval | 12.1 to 23.6 | 11.9 to 18.3 |
| hazard-ratio | ||
| (HYCAMTIN:paclitaxel) | 0.76 | |
| (p-value) | (0.07) | |
| Survival (weeks) | ||
| Median | 63.0 | 53.0 |
| 95% Confidence interval | 46.6 to 71.9 | 42.3 to 68.7 |
| hazard-ratio | ||
| (HYCAMTIN:paclitaxel) | 0.97 | |
| (p-value) | (0.87) | |
* The calculation for duration of response was based on the interval between first response and time to progression.
The median time to response was 7.6 weeks (range 3.1 to 21.7) with HYCAMTIN compared to 6.0 weeks (range 2.4 to 18.1) with paclitaxel. Consequently, the efficacy of HYCAMTIN may not be achieved if patients are withdrawn from treatment prematurely.
In the crossover phase, 8 of 61 (13%) patients who received HYCAMTIN after paclitaxel had a partial response and 5 of 49 (10%) patients who received paclitaxel after HYCAMTIN had a response (2 complete responses).
HYCAMTIN was active in ovarian cancer patients who had developed resistance to platinum-containing therapy, defined as tumor progression while on, or tumor relapse within 6 months after completion of, a platinum-containing regimen. One complete and 6 partial responses were seen in 60 patients, for a response rate of 12%. In the same study, there were no complete responders and 4 partial responders on the paclitaxel arm, for a response rate of 7%.
HYCAMTIN was also studied in an open-label, non-comparative trial in 111 patients with recurrent ovarian cancer after treatment with a platinum-containing regimen, or who had not responded to 1 prior platinum-containing regimen. The response rate was 14% (95% CI = 7% to 20%). The median duration of response was 22 weeks (range 4.6 to 41.9 weeks). The time to progression was 11.3 weeks (range 0.7 to 72.1 weeks). The median survival was 67.9 weeks (range 1.4 to 112.9 weeks).
Small Cell Lung Cancer
HYCAMTIN was studied in 426 patients with recurrent or progressive small cell lung cancer in 1 randomized, comparative study and in 3 single-arm studies.
Randomized Comparative Study
In a randomized, comparative, Phase 3 trial, 107 patients were treated with HYCAMTIN (1.5 mg/m2/day × 5 days starting on day 1 of a 21-day course) and 104 patients were treated with CAV (1,000 mg/m2 cyclophosphamide, 45 mg/m2 doxorubicin, 2 mg vincristine administered sequentially on day 1 of a 21-day course). All patients were considered sensitive to first-line chemotherapy (responders who then subsequently progressed≥60 days after completion of first-line therapy). A total of 77% of patients treated with HYCAMTIN and 79% of patients treated with CAV received platinum/etoposide with or without other agents as first-line chemotherapy.
Response rates, response duration, time to progression, and survival are shown in Table 2.
| Parameter |
HYCAMTIN (n = 107) |
CAV (n = 104) |
| Complete response rate | 0% | 1% |
| Partial response rate | 24% | 17% |
| Overall response rate | 24% | 18% |
| Difference in overall response rates | 6% | |
| 95% Confidence interval of the difference | (–6 to 18%) | |
| Response duration* (weeks) | n = 26 | n = 19 |
| Median | 14.4 | 15.3 |
| 95% Confidence interval | 13.1 to 18.0 | 13.1 to 23.1 |
| hazard-ratio | ||
| (HYCAMTIN:CAV) (95% CI) | 1.42 (0.73 to 2.76) | |
| (p-value) | (0.30) | |
| Time to progression (weeks) | ||
| Median | 13.3 | 12.3 |
| 95% Confidence interval | 11.4 to 16.4 | 11.0 to 14.1 |
| hazard-ratio | ||
| (HYCAMTIN:CAV) (95% CI) | 0.92 (0.69 to 1.22) | |
| (p-value) | (0.55) | |
| Survival (weeks) | ||
| Median | 25.0 | 24.7 |
| 95% Confidence interval | 20.6 to 29.6 | 21.7 to 30.3 |
| hazard-ratio | ||
| (HYCAMTIN:CAV) (95% CI) | 1.04 (0.78 to 1.39) | |
| (p-value) | (0.80) | |
* The calculation for duration of response was based on the interval between first response and time to progression.
The time to response was similar in both arms: HYCAMTIN median of 6 weeks (range 2.4 to 15.7) versus CAV median 6 weeks (range 5.1 to 18.1).
Changes on a disease-related symptom scale in patients who received HYCAMTIN or who received CAV are presented in Table 3. It should be noted that not all patients had all symptoms, nor did all patients respond to all questions. Each symptom was rated on a 4-category scale with an improvement defined as a change in 1 category from baseline sustained over 2 courses. Limitations in interpretation of the rating scale and responses preclude formal statistical analysis.
| Symptom |
HYCAMTIN (n = 107) |
CAV (n = 104) |
||
| n† | (%) | n† | (%) | |
| Shortness of breath | 68 | (28) | 61 | (7) |
| Interference with daily activity | 67 | (27) | 63 | (11) |
| Fatigue | 70 | (23) | 65 | (9) |
| Hoarseness | 40 | (33) | 38 | (13) |
| Cough | 69 | (25) | 61 | (15) |
| Insomnia | 57 | (33) | 53 | (19) |
| Anorexia | 56 | (32) | 57 | (16) |
| Chest pain | 44 | (25) | 41 | (17) |
| Hemoptysis | 15 | (27) | 12 | (33) |
* Defined as improvement sustained over at least 2 courses compared to baseline.
† Number of patients with baseline and at least 1 post-baseline assessment.
Single-Arm Studies
HYCAMTIN was also studied in 3 open-label, non-comparative trials in a total of 319 patients with recurrent or progressive small cell lung cancer after treatment with first-line chemotherapy. In all 3 studies, patients were stratified as either sensitive (responders who then subsequently progressed ≥90 days after completion of first-line therapy) or refractory (no response to first-line chemotherapy or who responded to first-line therapy and then progressed within 90 days of completing first-line therapy). Response rates ranged from 11% to 31% for sensitive patients and 2% to 7% for refractory patients. Median time to progression and median survival were similar in all 3 studies and the comparative study.
Cervical Cancer
In a comparative trial, 147 eligible women were randomized to HYCAMTIN (0.75 mg/m2/day IV over 30 minutes × 3 consecutive days starting on day 1 of a 21-day course) plus cisplatin (50 mg/m2 on day 1) and 146 eligible women were randomized to cisplatin (50 mg/m2 IV on day 1 of a 21-day course). All patients had histologically confirmed Stage IV-B, recurrent, or persistent carcinoma of the cervix considered not amenable to curative treatment with surgery and/or radiation. Fifty-six percent (56%) of patients treated with HYCAMTIN plus cisplatin and 56% of patients treated with cisplatin had received prior cisplatin with or without other agents as first-line chemotherapy.
Median survival of eligible patients in the HYCAMTIN plus cisplatin treatment arm was 9.4 months (95% CI: 7.9 to 11.9) compared to 6.5 months (95% CI: 5.8 to 8.8) among patients randomized to cisplatin alone with a log rank p-value of 0.033 (significance level was 0.044 after adjusting for the interim analysis). The unadjusted hazard ratio for overall survival was 0.76 (95% CI: 0.59 to 0.98).
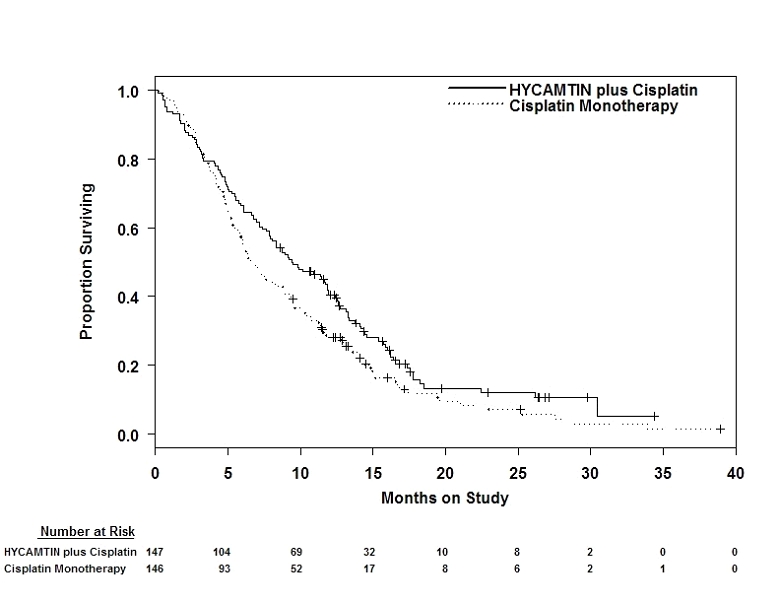
Figure 1. Overall Survival Curves Comparing HYCAMTIN plus Cisplatin versus Cisplatin Monotherapy in Cervical Cancer Patients
INDICATIONS AND USAGE
HYCAMTIN is indicated for the treatment of:
- metastatic carcinoma of the ovary after failure of initial or subsequent chemotherapy.
- small cell lung cancer sensitive disease after failure of first-line chemotherapy. In clinical studies submitted to support approval, sensitive disease was defined as disease responding to chemotherapy but subsequently progressing at least 60 days (in the Phase 3 study) or at least 90 days (in the Phase 2 studies) after chemotherapy (see CLINICAL STUDIES).
HYCAMTIN in combination with cisplatin is indicated for the treatment of:
- stage IV-B, recurrent, or persistent carcinoma of the cervix which is not amenable to curative treatment with surgery and/or radiation therapy.
CONTRAINDICATIONS
HYCAMTIN is contraindicated in patients who have a history of hypersensitivity reactions to topotecan or to any of its ingredients. HYCAMTIN should not be used in patients who are pregnant or breast-feeding, or those with severe bone marrow depression.
WARNINGS
Bone marrow suppression (primarily neutropenia) is the dose-limiting toxicity of HYCAMTIN. Neutropenia is not cumulative over time. The following data on myelosuppression is based on:
- the combined experience of 879 patients with metastatic ovarian cancer or small cell lung cancer treated with HYCAMTIN monotherapy at a dose of 1.5 mg/m2/day x 5 days.
- the experience of 140 patients with cervical cancer randomized to receive HYCAMTIN 0.75 mg/m2/day on days 1, 2, and 3 plus cisplatin 50 mg/m2 on day 1.
Neutropenia
- Ovarian and small cell lung cancer experience: Grade 4 neutropenia (<500 cells/mm3) was most common during course 1 of treatment (60% of patients) and occurred in 39% of all courses, with a median duration of 7 days. The nadir neutrophil count occurred at a median of 12 days. Therapy-related sepsis or febrile neutropenia occurred in 23% of patients, and sepsis was fatal in 1%. Pancytopenia has been reported.
- Topotecan-induced neutropenia can lead to neutropenic colitis. Fatalities due to neutropenic colitis have been reported in clinical trials with HYCAMTIN. In patients presenting with fever, neutropenia, and a compatible pattern of abdominal pain, the possibility of neutropenic colitis should be considered.
- Cervical cancer experience: Grade 3 and grade 4 neutropenia affected 26% and 48% of patients, respectively.
Thrombocytopenia
- Ovarian and small cell lung cancer experience: Grade 4 thrombocytopenia (<25,000/mm3) occurred in 27% of patients and in 9% of courses, with a median duration of 5 days and platelet nadir at a median of 15 days. Platelet transfusions were given to 15% of patients in 4% of courses.
- Cervical cancer experience: Grade 3 and grade 4 thrombocytopenia affected 26% and 7% of patients, respectively.
Anemia
- Ovarian and small cell lung cancer experience: Grade 3/4 anemia (<8 g/dL) occurred in 37% of patients and in 14% of courses. Median nadir was at day 15. Transfusions were needed in 52% of patients in 22% of courses.
- Cervical cancer experience: Grade 3 and grade 4 anemia affected 34% and 6% of patients, respectively.
In ovarian cancer, the overall treatment-related death rate was 1%. In the comparative study in small cell lung cancer, however, the treatment-related death rates were 5% for HYCAMTIN and 4% for CAV.
Monitoring of Bone Marrow Function
HYCAMTIN should be administered only in patients with adequate bone marrow reserves, including baseline neutrophil count of at least 1,500 cells/mm3 and platelet count at least 100,000/mm3. Frequent monitoring of peripheral blood cell counts should be instituted during treatment with HYCAMTIN. Patients should not be treated with subsequent courses of HYCAMTIN until neutrophils recover to >1,000 cells/mm3, platelets recover to >100,000 cells/mm3, and hemoglobin levels recover to 9.0 g/dL (with transfusion if necessary). Severe myelotoxicity has been reported when HYCAMTIN is used in combination with cisplatin (see Drug Interactions).
Interstitial Lung Disease
HYCAMTIN has been associated with reports of interstitial lung disease (ILD), some of which have been fatal (see ADVERSE REACTIONS). Underlying risk factors include history of ILD, pulmonary fibrosis, lung cancer, thoracic exposure to radiation, and use of pneumotoxic drugs and/or colony stimulating factors. Patients should be monitored for pulmonary symptoms indicative of interstitial lung disease (e.g., cough, fever, dyspnea, and/or hypoxia), and HYCAMTIN should be discontinued if a new diagnosis of ILD is confirmed.
Pregnancy
HYCAMTIN may cause fetal harm when administered to a pregnant woman. The effects of topotecan on pregnant women have not been studied. If topotecan is used during a patient’s pregnancy, or if a patient becomes pregnant while taking topotecan, she should be warned of the potential hazard to the fetus. Women should be warned to avoid becoming pregnant. (See CONTRAINDICATIONS.) In rabbits, a dose of 0.10 mg/kg/day (about equal to the clinical dose on a mg/m2 basis) given on days 6 through 20 of gestation caused maternal toxicity, embryolethality, and reduced fetal body weight. In the rat, a dose of 0.23 mg/kg/day (about equal to the clinical dose on a mg/m2 basis) given for 14 days before mating through gestation day 6 caused fetal resorption, microphthalmia, pre-implant loss, and mild maternal toxicity. A dose of 0.10 mg/kg/day (about half the clinical dose on a mg/m2 basis) given to rats on days 6 through 17 of gestation caused an increase in post-implantation mortality. This dose also caused an increase in total fetal malformations. The most frequent malformations were of the eye (microphthalmia, anophthalmia, rosette formation of the retina, coloboma of the retina, ectopic orbit), brain (dilated lateral and third ventricles), skull, and vertebrae.
PRECAUTIONS
General
Inadvertent extravasation with HYCAMTIN has been associated only with mild local reactions such as erythema and bruising.
Information for Patients
As with other chemotherapeutic agents, HYCAMTIN may cause asthenia or fatigue; if these symptoms occur, caution should be observed when driving or operating machinery.
Hematology
Monitoring of bone marrow function is essential (see WARNINGS and DOSAGE AND ADMINISTRATION).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity testing of topotecan has not been performed. Topotecan, however, is known to be genotoxic to mammalian cells and is a probable carcinogen. Topotecan was mutagenic to L5178Y mouse lymphoma cells and clastogenic to cultured human lymphocytes with and without metabolic activation. It was also clastogenic to mouse bone marrow. Topotecan did not cause mutations in bacterial cells.
Drug Interactions
Concomitant administration of G-CSF can prolong the duration of neutropenia, so if G-CSF is to be used, it should not be initiated until day 6 of the course of therapy, 24 hours after completion of treatment with HYCAMTIN.
Myelosuppressionwas more severe when HYCAMTIN, at a dose of 1.25 mg/m2/day × 5 days, was given in combination with cisplatin at a dose of 50 mg/m2 in Phase 1 studies. In one study, 1 of 3 patients had severe neutropenia for 12 days and a second patient died with neutropenic sepsis.
Greater myelosuppression is also likely to be seen when HYCAMTIN is used in combination with other cytotoxic agents, thereby necessitating a dose reduction. However, when combining HYCAMTIN with platinum agents (e.g., cisplatin or carboplatin), a distinct sequence-dependent interaction on myelosuppression has been reported. Coadministration of a platinum agent on day 1 of HYCAMTIN dosing required lower doses of each agent compared to coadministration on day 5 of the HYCAMTIN dosing schedule.
For information on the pharmacokinetics, efficacy, safety, and dosing of HYCAMTIN at a dose of 0.75 mg/m2/day on days 1, 2, and 3 in combination with cisplatin 50 mg/m2 on day 1 for cervical cancer, see CLINICAL PHARMACOLOGY, CLINICAL STUDIES, ADVERSE REACTIONS, and DOSAGE AND ADMINISTRATION.
Pregnancy
Pregnancy Category D. (See WARNINGS.)
Nursing Mothers
It is not known whether the drug is excreted in human milk. Breastfeeding should be discontinued when women are receiving HYCAMTIN (see CONTRAINDICATIONS).
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Of the 879 patients with metastatic ovarian cancer or small cell lung cancer in clinical studies of HYCAMTIN, 32% (n = 281) were 65 years of age and older, while 3.8% (n = 33) were 75 years of age and older. Of the 140 patients with stage IV-B, relapsed, or refractory cervical cancer in clinical studies of HYCAMTIN who received HYCAMTIN plus cisplatin in the randomized clinical trial, 6% (n = 9) were 65 years of age and older, while 3% (n = 4) were 75 years of age and older. No overall differences in effectiveness or safety were observed between these patients and younger adult patients, and other reported clinical experience has not identified differences in responses between the elderly and younger adult patients, but greater sensitivity of some older individuals cannot be ruled out.
There were no apparent differences in the pharmacokinetics of topotecan in elderly patients, once the age-related decrease in renal function was considered (see CLINICAL PHARMACOLOGY).
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (see DOSAGE AND ADMINISTRATION).
ADVERSE REACTIONS
Ovarian Cancer and Small Cell Lung Cancer
Data in the following section are based on the combined experience of 453 patients with metastatic ovarian carcinoma, and 426 patients with small cell lung cancer treated with HYCAMTIN. Table 4 lists the principal hematologic toxicities, and Table 5 lists non-hematologic toxicities occurring in at least 15% of patients.
| Hematologic Adverse Event |
Patients n = 879 % Incidence |
Courses n = 4124 % Incidence |
| Neutropenia | ||
| <1,500 cells/mm3 | 97 | 81 |
| <500 cells/mm3 | 78 | 39 |
| Leukopenia | ||
| <3,000 cells/mm3 | 97 | 80 |
| <1,000 cells/mm3 | 32 | 11 |
| Thrombocytopenia | ||
| <75,000/mm3 | 69 | 42 |
| <25,000/mm3 | 27 | 9 |
| Anemia | ||
| <10 g/dL | 89 | 71 |
| <8 g/dL | 37 | 14 |
| Platelet transfusions | 15 | 4 |
| RBC transfusions | 52 | 22 |
|
Non-hematologic Adverse Event |
All Grades % Incidence |
Grade 3 % Incidence |
Grade 4 % Incidence |
|||
| n = 879 | n = 4124 | n = 879 | n = 4124 | n = 879 | n = 4124 | |
| Patients | Courses | Patients | Courses | Patients | Courses | |
| Infections and infestations | ||||||
| Sepsis or pyrexia/infection with neutropenia* | 43 | 15 | NR | NR | 23 | 7 |
| Metabolism and nutrition disorders | ||||||
| Anorexia | 19 | 9 | 2 | 1 | <1 | <1 |
| Nervous system disorders | ||||||
| Headache | 18 | 7 | 1 | <1 | <1 | 0 |
| Respiratory, thoracic, and mediastinal disorders | ||||||
| Dyspnea | 22 | 11 | 5 | 2 | 3 | 1 |
| Coughing | 15 | 7 | 1 | <1 | 0 | 0 |
| Gastrointestinal disorders | ||||||
| Nausea | 64 | 42 | 7 | 2 | 1 | <1 |
| Vomiting | 45 | 22 | 4 | 1 | 1 | <1 |
| Diarrhea | 32 | 14 | 3 | 1 | 1 | <1 |
| Constipation | 29 | 15 | 2 | 1 | 1 | <1 |
| Abdominal pain | 22 | 10 | 2 | 1 | 2 | <1 |
| Stomatitis | 18 | 8 | 1 | <1 | <1 | <1 |
| Skin and subcutaneous tissue disorders | ||||||
| Alopecia | 49 | 54 | NA | NA | NA | NA |
| Rash† | 16 | 6 | 1 | <1 | 0 | 0 |
| General disorders and administrative site conditions | ||||||
| Fatigue | 29 | 22 | 5 | 2 | 0 | 0 |
| Pyrexia | 28 | 11 | 1 | <1 | <1 | <1 |
| Pain‡ | 23 | 11 | 2 | 1 | 1 | <1 |
| Asthenia | 25 | 13 | 4 | 1 | 2 | <1 |
NA = Not applicable
NR = Not reported separately
* Does not include Grade 1 sepsis or pyrexia.
† Rash also includes pruritus, rash erythematous, urticaria, dermatitis, bullous eruption, and maculopapular rash.
‡ Pain includes body pain, back pain, and skeletal pain.
Premedications were not routinely used in these clinical studies.
Hematologic
(See WARNINGS.)
Nervous System Disorders
Headache (18% of patients) was the most frequently reported neurologic toxicity. Paresthesia occurred in 7% of patients but was generally grade 1.
Respiratory, Thoracic, and Mediastinal Disorders
The incidence of grade 3/4 dyspnea was 4% in ovarian cancer patients and 12% in small cell lung cancer patients.
Gastrointestinal Disorders
The incidence of nausea was 64% (8% grade 3/4), and vomiting occurred in 45% (6% grade 3/4) of patients (see Table 5). The prophylactic use of antiemetics was not routine in patients treated with HYCAMTIN. Thirty-two percent of patients had diarrhea (4% grade 3/4), 29% constipation (2% grade 3/4), and 22% had abdominal pain (4% grade 3/4). Grade 3/4 abdominal pain was 6% in ovarian cancer patients and 2% in small cell lung cancer patients.
Skin and Subcutaneous Tissue Disorders
Total alopecia (grade 2) occurred in 31% of patients.
Hepatobiliary Disorders
Grade 1 transient elevations in hepatic enzymes occurred in 8% of patients. Greater elevations, grade 3/4, occurred in 4%. Grade 3/4 elevated bilirubin occurred in <2% of patients.
Table 6 shows the grade 3/4 hematologic and major non-hematologic adverse events in the topotecan/paclitaxel comparator trial in ovarian cancer.
| Adverse Event | HYCAMTIN | Paclitaxel | ||
| Patients | Courses | Patients | Courses | |
| n = 112 | n = 597 | n = 114 | n = 589 | |
| Hematologic Grade 3/4 | % | % | % | % |
|
Grade 4 neutropenia (<500 cells/mm3) | 80 | 36 | 21 | 9 |
|
Grade 3/4 anemia (Hgb <8 g/dL) | 41 | 16 | 6 | 2 |
|
Grade 4 thrombocytopenia (<25,000 plts/mm3) | 27 | 10 | 3 | <1 |
| Pyrexia/Grade 4 neutropenia | 23 | 6 | 4 | 1 |
| Non-hematologic Grade 3/4 | % | % | % | % |
| Infections and infestations | ||||
| Documented sepsis | 5 | 1 | 2 | <1 |
| Death related to sepsis | 2 | NA | 0 | NA |
| Metabolism and nutrition disorders | ||||
| Anorexia | 4 | 1 | 0 | 0 |
| Nervous system disorders | ||||
| Headache | 1 | <1 | 2 | 1 |
| Respiratory, thoracic, and mediastinal disorders | ||||
| Dyspnea | 6 | 2 | 5 | 1 |
| Gastrointestinal disorders | ||||
| Abdominal pain | 5 | 1 | 4 | 1 |
| Constipation | 5 | 1 | 0 | 0 |
| Diarrhea | 6 | 2 | 1 | <1 |
| Intestinal obstruction | 5 | 1 | 4 | 1 |
| Nausea | 10 | 3 | 2 | <1 |
| Stomatitis | 1 | <1 | 1 | <1 |
| Vomiting | 10 | 2 | 3 | <1 |
| Hepatobiliary Disorders | ||||
| Hepatic enzymes increased* | 1 | <1 | 1 | <1 |
| Skin and subcutaneous tissue disorders | ||||
| Rash† | 0 | 0 | 1 | <1 |
| Musculoskeletal, connective tissue, and bone disorders | ||||
| Arthralgia | 1 | <1 | 3 | <1 |
| General disorders and administrative site conditions | ||||
| Fatigue | 7 | 2 | 6 | 2 |
| Malaise | 2 | <1 | 2 | <1 |
| Asthenia | 5 | 2 | 3 | 1 |
| Chest pain | 2 | <1 | 1 | <1 |
| Myalgia | 0 | 0 | 3 | 2 |
| Pain‡ | 5 | 1 | 7 | 2 |
* Increased hepatic enzymes includes increased SGOT/AST, increased SGPT/ALT, and increased hepatic enzymes.
† Rash also includes pruritus, rash erythematous, urticaria, dermatitis, bullous eruption, and maculopapular rash.
‡ Pain includes body pain, skeletal pain, and back pain.
Premedications were not routinely used in patients randomized to HYCAMTIN, whereas patients receiving paclitaxel received routine pretreatment with corticosteroids, diphenhydramine, and histamine receptor type 2 blockers.
Table 7 shows the grade 3/4 hematologic and major non-hematologic adverse events in the topotecan/CAV comparator trial in small cell lung cancer.
| Adverse Event | HYCAMTIN | CAV | ||
| Patients | Courses | Patients | Courses | |
| n = 107 | n = 446 | n = 104 | n = 359 | |
| Hematologic Grade 3/4 | % | % | % | % |
|
Grade 4 neutropenia (<500 cells/mm3) | 70 | 38 | 72 | 51 |
|
Grade 3/4 anemia (Hgb <8 g/dL) | 42 | 18 | 20 | 7 |
|
Grade 4 thrombocytopenia (<25,000 plts/mm3) | 29 | 10 | 5 | 1 |
| Pyrexia/Grade 4 neutropenia | 28 | 9 | 26 | 13 |
| Non-hematologic Grade 3/4 | % | % | % | % |
| Infections and infestations | ||||
| Documented sepsis | 5 | 1 | 5 | 1 |
| Death related to sepsis | 3 | NA | 1 | NA |
| Metabolism and nutrition disorders | ||||
| Anorexia | 3 | 1 | 4 | 2 |
| Nervous system disorders | ||||
| Headache | 0 | 0 | 2 | <1 |
| Respiratory, thoracic, and mediastinal disorders | ||||
| Dyspnea | 9 | 5 | 14 | 7 |
| Coughing | 2 | 1 | 0 | 0 |
| Pneumonia | 8 | 2 | 6 | 2 |
| Gastrointestinal disorders | ||||
| Abdominal pain | 6 | 1 | 4 | 2 |
| Constipation | 1 | <1 | 0 | 0 |
| Diarrhea | 1 | <1 | 0 | 0 |
| Nausea | 8 | 2 | 6 | 2 |
| Stomatitis | 2 | <1 | 1 | <1 |
| Vomiting | 3 | <1 | 3 | 1 |
| Hepatobiliary Disorders | ||||
| Increased hepatic enzymes* | 1 | <1 | 0 | 0 |
| Skin and subcutaneous tissue disorders | ||||
| Rash† | 1 | <1 | 1 | <1 |
| General disorders and administrative site conditions | ||||
| Fatigue | 6 | 4 | 10 | 3 |
| Asthenia | 9 | 4 | 7 | 2 |
| Pain‡ | 5 | 2 | 7 | 4 |
* Increased hepatic enzymes includes increased SGOT/AST, increased SGPT/ALT, and increased hepatic enzymes.
† Rash also includes pruritus, rash erythematous, urticaria, dermatitis, bullous eruption, and maculopapular rash.
‡ Pain includes body pain, skeletal pain, and back pain.
Premedications were not routinely used in patients randomized to HYCAMTIN, whereas patients receiving CAV received routine pretreatment with corticosteroids, diphenhydramine, and histamine receptor type 2 blockers.
Cervical Cancer
In the HYCAMTIN plus cisplatin versus cisplatin comparative trial in cervical cancer patients, the most common dose-limiting toxicity was myelosuppression. Table 8 shows the hematologic adverse events and Table 9 shows the non-hematologic adverse events in cervical cancer patients.
| Hematologic Adverse Event | HYCAMTIN Plus Cisplatin (n = 140) |
Cisplatin (n = 144) |
| Anemia | ||
| All grades (Hgb <12 g/dL) | 131 (94%) | 130 (90%) |
| Grade 3 (Hgb <8-6.5 g/dL) | 47 (34%) | 28 (19%) |
| Grade 4 (Hgb <6.5 g/dL) | 9 (6%) | 5 (3%) |
| Leukopenia | ||
| All grades (<3,800 cells/mm3) | 128 (91%) | 43 (30%) |
| Grade 3 (<2,000-1,000 cells/mm3) | 58 (41%) | 1 (1%) |
| Grade 4 (<1,000 cells/mm3) | 35 (25%) | 0 (0%) |
| Neutropenia | ||
| All grades (<2,000 cells/mm3) | 125 (89%) | 28 (19%) |
| Grade 3 (<1,000-500 cells/mm3) | 36 (26%) | 1 (1%) |
| Grade 4 (<500 cells/mm3) | 67 (48%) | 1 (1%) |
| Thrombocytopenia | ||
| All grades (<130,000 cells/mm3) | 104 (74%) | 21 (15%) |
| Grade 3 (<50,000-10,000 cells/mm3) | 36 (26%) | 5 (3%) |
| Grade 4 (<10,000 cells/mm3) | 10 (7%) | 0 (0%) |
* Includes patients who were eligible and treated.
| HYCAMTIN Plus Cisplatin | Cisplatin | |||||
| n = 140 | n = 144 | |||||
| Adverse Event | All Grades† | Grade 3 | Grade 4 | All Grades† | Grade 3 | Grade 4 |
| General disorders and administrative site conditions | ||||||
| Constitutional‡ | 96 (69%) | 11 (8%) | 0 | 89 (62%) | 17 (12%) | 0 |
| Pain§ | 82 (59%) | 28 (20%) | 3 (2%) | 72 (50%) | 18 (13%) | 5 (3%) |
| Gastrointestinal disorders | ||||||
| Vomiting | 56 (40%) | 20 (14%) | 2 (1%) | 53 (37%) | 13 (9%) | 0 |
| Nausea | 77 (55%) | 18 (13%) | 2 (1%) | 79 (55%) | 13 (9%) | 0 |
| Stomatitis-pharyngitis | 8 (6%) | 1 (<1%) | 0 | 0 | 0 | 0 |
| Other | 88 (63%) | 16 (11%) | 4 (3%) | 80 (56%) | 12 (8%) | 3 (2%) |
| Dermatology | 67 (48%) | 1 (<1%) | 0 | 29 (20%) | 0 | 0 |
| Metabolic-Laboratory | 55 (39%) | 13 (9%) | 7 (5%) | 44 (31%) | 14 (10%) | 1 (<1%) |
| Genitourinary | 51 (36%) | 9 (6%) | 9 (6%) | 49 (34%) | 7 (5%) | 7 (5%) |
| Nervous system disorders | ||||||
| Neuropathy | 4 (3%) | 1 (<1%) | 0 | 3 (2%) | 1 (<1%) | 0 |
| Other | 49 (35%) | 3 (2%) | 1 (<1%) | 43 (30%) | 7 (5%) | 2 (1%) |
| Infection-febrile neutropenia | 39 (28%) | 21 (15%) | 5 (4%) | 26 (18%) | 11 (8%) | 0 |
| Cardiovascular | 35 (25%) | 7 (5%) | 6 (4%) | 22 (15%) | 8 (6%) | 3 (2%) |
| Hepatic | 34 (24%) | 5 (4%) | 2 (1%) | 23 (16%) | 2 (1%) | 0 |
| Pulmonary | 24 (17%) | 4 (3%) | 0 | 23 (16%) | 5 (3%) | 3 (2%) |
| Vascular disorders | ||||||
| Hemorrhage | 21 (15%) | 8 (6%) | 1 (<1%) | 20 (14%) | 3 (2%) | 1 (<1%) |
| Coagulation | 8 (6%) | 4 (3%) | 3 (2%) | 10 (7%) | 7 (5%) | 0 |
| Musculoskeletal | 19 (14%) | 3 (2%) | 0 | 7 (5%) | 1 (<1%) | 1 (<1%) |
| Allergy-Immunology | 8 (6%) | 2 (1%) | 1 (<1%) | 4 (3%) | 0 | 1 (<1%) |
| Endocrine | 8 (6%) | 0 | 0 | 4 (3%) | 2 (1%) | 0 |
| Sexual reproduction function | 7 (5%) | 0 | 0 | 10 (7%) | 1 (<1%) | 0 |
| Ocular-visual | 7 (5%) | 0 | 0 | 7 (5%) | 1 (<1%) | 0 |
Data were collected using NCI Common Toxicity Criteria, v. 2.0.
* Includes patients who were eligible and treated.
† Grades 1 through 4 only. There were 3 patients who experienced grade 5 deaths with investigator-designated attribution. One was a grade 5 hemorrhage in which the drug-related thrombocytopenia aggravated the event. A second patient experienced bowel obstruction, cardiac arrest, pleural effusion and respiratory failure which were not treatment related but probably aggravated by treatment. A third patient experienced a pulmonary embolism and adult respiratory distress syndrome, the latter was indirectly treatment-related.
‡ Constitutional includes fatigue (lethargy, malaise, asthenia), fever (in the absence of neutropenia), rigors, chills, sweating, and weight gain or loss.
§ Pain includes abdominal pain or cramping, arthralgia, bone pain, chest pain (non-cardiac and non-pleuritic), dysmenorrhea, dyspareunia, earache, headache, hepatic pain, myalgia, neuropathic pain, pain due to radiation, pelvic pain, pleuritic pain, rectal or perirectal pain, and tumor pain.
Postmarketing Reports of Adverse Events
Reports of adverse events in patients taking HYCAMTIN received after market introduction, which are not listed above, include the following:
Blood and Lymphatic System Disorders
Rare: Severe bleeding (in association with thrombocytopenia).
Immune System Disorders
Infrequent: Allergic manifestations; rare: Anaphylactoid reactions.
Gastrointestinal Disorders
Abdominal pain potentially associated with neutropenic colitis (see WARNINGS).
Pulmonary Disorders
Interstitial lung disease.
Skin and Subcutaneous Tissue Disorders
Rare: Angioedema, severe dermatitis, severe pruritus.
OVERDOSAGE
There is no known antidote for overdosage with HYCAMTIN. The primary anticipated complication of overdosage would consist of bone marrow suppression.
One patient on a single-dose regimen of 17.5 mg/m2 given on day 1 of a 21-day cycle had received a single dose of 35 mg/m2. This patient experienced severe neutropenia (nadir of 320/mm3) 14 days later but recovered without incident.
The LD10 in mice receiving single intravenous infusions of HYCAMTIN was 75 mg/m2 (CI 95%: 47 to 97).
DOSAGE AND ADMINISTRATION
Ovarian Cancer and Small Cell Lung Cancer
Prior to administration of the first course of HYCAMTIN, patients must have a baseline neutrophil count of >1,500 cells/mm3 and a platelet count of >100,000 cells/mm3. The recommended dose of HYCAMTIN is 1.5 mg/m2 by intravenous infusion over 30 minutes daily for 5 consecutive days, starting on day 1 of a 21-day course.
In the absence of tumor progression, a minimum of 4 courses is recommended because tumor response may be delayed. The median time to response in 3 ovarian clinical trials was 9 to 12 weeks, and median time to response in 4 small cell lung cancer trials was 5 to 7 weeks.
In the event of severe neutropenia during any course, the dose should be reduced by 0.25 mg/m2 (to 1.25 mg/m2) for subsequent courses. Doses should be similarly reduced if the platelet count falls below 25,000 cells/mm3. Alternatively, in the event of severe neutropenia, G-CSF may be administered following the subsequent course (before resorting to dose reduction) starting from day 6 of the course (24 hours after completion of topotecan administration).
Cervical Cancer
Prior to administration of the first course of HYCAMTIN, patients must have a baseline absolute neutrophil count of >1,500 cells/mm3 and a platelet count of >100,000 cells/mm3. The recommended dose of HYCAMTIN is 0.75 mg/m2 by intravenous infusion over 30 minutes daily on days 1, 2, and 3; followed by cisplatin 50 mg/m2 by intravenous infusion on day 1 repeated every 21 days (a 21-day course).
Dosage adjustments for subsequent courses of HYCAMTIN in combination with cisplatin are specific for each drug.
- In the event of severe febrile neutropenia (defined as <1,000 cells/mm3 with temperature of 38.0°C or 100.4°F), the dose of HYCAMTIN should be reduced by 20% to 0.60 mg/m2 for subsequent courses. Doses of HYCAMTIN should be similarly reduced (by 20% to 0.60 mg/m2) if the platelet count falls below 10,000 cells/mm3. Alternatively, in the event of severe febrile neutropenia, G-CSF may be administered following the subsequent course (before resorting to dose reduction) starting from day 4 of the course (24 hours after completion of administration of HYCAMTIN). If febrile neutropenia occurs despite the use of G-CSF, the dose of HYCAMTIN should be reduced by another 20% to 0.45 mg/m2 for subsequent courses.
- See manufacturer’s prescribing information for cisplatin administration and hydration guidelines and for cisplatin dosage adjustment in the event of hematologic toxicity.
Adjustment of Dose in Special Populations
Hepatic Impairment
No dosage adjustment appears to be required for treating patients with impaired hepatic function (plasma bilirubin >1.5 to <10 mg/dL).
Renal Functional Impairment
No dosage adjustment of HYCAMTIN appears to be required for treating patients with mild renal impairment (Clcr 40 to 60 mL/min.). Dosage adjustment of HYCAMTIN to 0.75 mg/m2 is recommended for patients with moderate renal impairment (20 to 39 mL/min.). Insufficient data are available in patients with severe renal impairment to provide a dosage recommendation for HYCAMTIN.
HYCAMTIN in combination with cisplatin for the treatment of cervical cancer should only be initiated in patients with serum creatinine≤1.5 mg/dL. In the clinical trial, cisplatin was discontinued for a serum creatinine >1.5 mg/dL. Insufficient data are available regarding continuing monotherapy with HYCAMTIN after cisplatin discontinuation in patients with cervical cancer.
Elderly Patients
No dosage adjustment appears to be needed in the elderly other than adjustments related to renal function (see CLINICAL PHARMACOLOGY and PRECAUTIONS).
PREPARATION FOR ADMINISTRATION
Precautions
HYCAMTIN is a cytotoxic anticancer drug. As with other potentially toxic compounds, HYCAMTIN should be prepared under a vertical laminar flow hood while wearing gloves and protective clothing. If HYCAMTIN solution contacts the skin, wash the skin immediately and thoroughly with soap and water. If HYCAMTIN contacts mucous membranes, flush thoroughly with water.
Preparation for Intravenous Administration
Each HYCAMTIN 4-mg vial is reconstituted with 4 mL Sterile Water for Injection. Then the appropriate volume of the reconstituted solution is diluted in either 0.9% Sodium Chloride Intravenous Infusion or 5% Dextrose Intravenous Infusion prior to administration.
Because the lyophilized dosage form contains no antibacterial preservative, the reconstituted product should be used immediately.
STABILITY
Unopened vials of HYCAMTIN are stable until the date indicated on the package when stored between 20° and 25°C (68° and 77°F) [see USP] and protected from light in the original package. Because the vials contain no preservative, contents should be used immediately after reconstitution.
Reconstituted vials of HYCAMTIN diluted for infusion are stable at approximately 20° to 25°C (68° to 77°F) and ambient lighting conditions for 24 hours.
HOW SUPPLIED
HYCAMTIN for Injection is supplied in 4-mg (free base) single-dose vials.
NDC 0007-4201-01 (package of 1)
NDC 0007-4201-05 (package of 5)
Storage
Store the vials protected from light in the original cartons at controlled room temperature between 20° and 25°C (68° and 77°F) [see USP].
Handling and Disposal
Procedures for proper handling and disposal of anticancer drugs should be used. Several guidelines on this subject have been published.1-8 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
REFERENCES
- Brown KA, Esper P, Kelleher LO, Brace O'Neill JE, Polovich M, White JM, eds. In: Chemotherapy and Biotherapy Guidelines and Recommendations for Practice. Pittsburgh, PA: Oncology Nursing Society:2001:55-73.
- National Institutes of Health Web site. Recommendations for the safe handling of cytotoxic drugs. NIH Publication 92-2621. Available at http://www.nih.gov/od/ors/ds/pubs/cyto/index.htm. Accessed August 21, 2002.
- AMA Council on Scientific Affairs. Guidelines for handling parenteral antineoplastics. JAMA 1985;253(11):1590-1591.
- National Study Commission on Cytotoxic Exposure — Recommendations for handling cytotoxic agents. 1987. Available from Louis P. Jeffrey, Sc.D., Chairman, National Study Commission on Cytotoxic Exposure. Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, MA 02115.
- Clinical Oncological Society of Australia. Guidelines and recommendations for safe handling of antineoplastic agents. Med J Austr 1983;1:426-428.
- Jones RB, Frank R, Mass T. Safe handling of chemotherapeutic agents: A report from the Mount Sinai Medical Center. CA-A Cancer J for Clin 1983;33:258-263.
- American Society of Hospital Pharmacists. ASHP Technical Assistance Bulletin on Handling Cytotoxic and Hazardous Drugs. Am J Hosp Pharm 1990;47:1033-1049.
- Controlling Occupational Exposure to Hazardous Drugs. (OSHA Work-Practice Guidelines), Am J Health-Syst Pharm 1996;53:1669-1685.
GlaxoSmithKline
Research Triangle Park, NC 27709
HYCAMTIN is a registered trademark of GlaxoSmithKline.
©2009, GlaxoSmithKline. All rights reserved.
February 2009 HYJ:19PI
Principal Display Panel
NDC 0007-4201-01
HYCAMTIN®
TOPOTECAN HYDROCHLORIDE
FOR INJECTION
4 mg
For Intravenous Use
Rx only
Store at controlled room temperature between 20o and 25oC (68o and 77oF) [see USP]. Protect from light; product is light-sensitive.
Dosage: See accompanying prescribing information. Hycamtin is cytotoxic. Safe handling: Use caution in handling. See prescribing information for additional precautions. If solution contacts the skin, wash thoroughly with soap and water. Flush mucous membranes thoroughly with water.
GlaxoSmithKline
Research Triangle Park, NC 27709
10000000023714

| HYCAMTIN
topotecan hydrochloride injection, powder, lyophilized, for solution |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020671 | 06/07/1996 | |
| Labeler - SmithKline Beecham Corporation (167380711) |