BONIVA
-
ibandronate sodium injection, solution
Hoffmann-La Roche Inc
----------
BONIVA®(ibandronate sodium)
INJECTION
DESCRIPTION
BONIVA (ibandronate sodium) is a nitrogen-containing bisphosphonate that inhibits osteoclast-mediated bone resorption. The chemical name for ibandronate sodium is 3-(N-methyl-N-pentyl)amino-1-hydroxypropane-1,1-diphosphonic acid, monosodium salt, monohydrate with the molecular formula C9H22NO7P2Na•H2O and a molecular weight of 359.24. Ibandronate sodium is a white- to off-white powder. It is freely soluble in water and practically insoluble in organic solvents. Ibandronate sodium has the following structural formula:
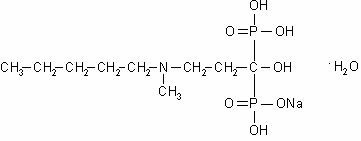
BONIVA Injection is intended for intravenous administration only. BONIVA Injection is available as a sterile, clear, colorless, ready-to-use solution in a prefilled syringe that delivers 3.375 mg of ibandronate monosodium salt monohydrate in 3 mL of solution, equivalent to a dose of 3 mg ibandronate free acid. Inactive ingredients include sodium chloride, glacial acetic acid, sodium acetate and water.
CLINICAL PHARMACOLOGY
Mechanism of Action
The action of ibandronate on bone tissue is based on its affinity for hydroxyapatite, which is part of the mineral matrix of bone. Ibandronate inhibits osteoclast activity and reduces bone resorption and turnover. In postmenopausal women, it reduces the elevated rate of bone turnover, leading to, on average, a net gain in bone mass.
Pharmacokinetics
Distribution
Area under the serum ibandronate concentrations versus time curve increases in a dose-proportional manner after administration of 2 mg to 6 mg by intravenous injection. After administration, ibandronate either rapidly binds to bone or is excreted into urine. In humans, the apparent terminal volume of distribution is at least 90 L, and the amount of dose removed from the circulation into the bone is estimated to be 40% to 50% of the circulating dose. In vitro protein binding in human serum was approximately 86% over an ibandronate concentration range of 20 to 2000 ng/mL (approximate range of maximum serum ibandronate concentrations upon intravenous bolus administration) in one study.
Metabolism
There is no evidence that ibandronate is metabolized in humans. Ibandronate does not inhibit human P450 1A2, 2A6, 2C9, 2C19, 2D6, 2E1, and 3A4 isozymes in vitro.
Elimination
The portion of ibandronate that is not removed from the circulation via bone absorption is eliminated unchanged by the kidney (approximately 50% to 60% of the administered intravenous dose).
The plasma elimination of ibandronate is multiphasic. Its renal clearance and distribution into bone accounts for a rapid and early decline in plasma concentrations, reaching 10% of Cmax within 3 or 8 hours after intravenous or oral administration, respectively. This is followed by a slower clearance phase as ibandronate redistributes back into the blood from bone. The observed apparent terminal half-life for ibandronate is generally dependent on the dose studied and on assay sensitivity. The observed apparent terminal half-life for intravenous 2 and 4 mg ibandronate after 2 hours of infusion ranges from 4.6 to 15.3 hours and 5 to 25.5 hours, respectively.
Following intravenous administration, total clearance of ibandronate is low, with average values in the range 84 to 160 mL/min. Renal clearance (about 60 mL/min in healthy postmenopausal women) accounts for 50% to 60% of total clearance and is related to creatinine clearance. The difference between the apparent total and renal clearances likely reflects bone uptake of the drug.
Special Populations
Pediatrics
The pharmacokinetics of ibandronate has not been studied in patients <18 years of age.
Gender
The pharmacokinetics of ibandronate is similar in both men and women.
Geriatric
Since ibandronate is not known to be metabolized, the only difference in ibandronate elimination for geriatric patients versus younger patients is expected to relate to progressive age-related changes in renal function (see Special Populations: Renal Impairment).
Race
Pharmacokinetic differences due to race have not been studied.
Renal Impairment
Renal clearance of ibandronate in patients with various degrees of renal impairment is linearly related to creatinine clearance (CLcr).
Following a single dose of 0.5 mg ibandronate by intravenous administration, patients with CLcr 40 to 70 mL/min had 55% higher exposure (AUC∞) than the exposure observed in subjects with CLcr >90 mL/min. Patients with CLcr <30 mL/min had more than a two-fold increase in exposure compared to the exposure for healthy subjects (see DOSAGE AND ADMINISTRATION: Patients with Renal Impairment).
Hepatic Impairment
No studies have been performed to assess the pharmacokinetics of ibandronate in patients with hepatic impairment since ibandronate is not metabolized in the human liver.
Drug Interactions
Ibandronate does not undergo hepatic metabolism and does not inhibit the hepatic cytochrome P450 system. Ibandronate is eliminated by renal excretion. Based on a rat study, the ibandronate secretory pathway does not appear to include known acidic or basic transport systems involved in the excretion of other drugs.
Melphalan/Prednisolone
A pharmacokinetic interaction study in multiple myeloma patients demonstrated that intravenous melphalan (10 mg/m2) and oral prednisolone (60 mg/m2) did not interact with 6 mg ibandronate upon intravenous coadministration. Ibandronate did not interact with melphalan or prednisolone.
Tamoxifen
A pharmacokinetic interaction study in healthy postmenopausal women demonstrated that there was no interaction between oral 30 mg tamoxifen and intravenous 2 mg ibandronate.
Pharmacodynamics
Osteoporosis is characterized by decreased bone mass and increased fracture risk, most commonly at the spine, hip, and wrist. The diagnosis can be confirmed by a finding of low bone mass, evidence of fracture on x-ray, a history of osteoporotic fracture, or height loss or kyphosis indicative of vertebral fracture. While osteoporosis occurs in both men and women, it is most common among women following menopause. In healthy humans, bone formation and resorption are closely linked; old bone is resorbed and replaced by newly formed bone. In postmenopausal osteoporosis, bone resorption exceeds bone formation, leading to bone loss and increased risk of fracture. After menopause, the risk of fractures of the spine and hip increases; approximately 40% of 50-year-old women will experience an osteoporosis-related fracture during their remaining lifetimes.
In studies of postmenopausal women, BONIVA Injection at doses of 0.5 mg to 3 mg produced biochemical changes indicative of inhibition of bone resorption, including decreases of biochemical markers of bone collagen degradation (cross-linked C-telopeptide of Type I collagen [CTX]). Changes in markers of bone formation (osteocalcin) were observed later than changes in resorption markers, as expected, due to the coupled nature of bone resorption and formation.
Year 1 results from an efficacy and safety study comparing BONIVA Injection 3 mg every 3 months and BONIVA 2.5 mg daily oral tablet demonstrated that both dosing regimens significantly suppressed serum CTX levels at Months 3, 6, and 12. The median pre-dose or trough serum CTX levels in the ITT population reached a nadir of 57% (BONIVA Injection) and 62% (BONIVA 2.5 mg tablets) below baseline values by Month 6, and remained stable at Month 12 of treatment.
Clinical Studies
Daily Oral Tablets
The effectiveness and safety of BONIVA daily oral tablets were demonstrated in a randomized, double-blind, placebo-controlled, multinational study (Treatment Study) of 2946 women aged 55 to 80 years, who were on average 21 years postmenopause, who had lumbar spine bone mineral density (BMD) 2 to 5 SD below the premenopausal mean (T-score) in at least one vertebra [L1-L4], and who had one to four prevalent vertebral fractures. BONIVA was evaluated at oral doses of 2.5 mg daily and 20 mg intermittently. The main outcome measure was the occurrence of new radiographically diagnosed, vertebral fractures after 3 years of treatment. The diagnosis of an incident vertebral fracture was based on both qualitative diagnosis by the radiologist and quantitative morphometric criterion. The morphometric criterion required the dual occurrence of two events: a relative height ratio or relative height reduction in a vertebral body of at least 20%, together with at least a 4 mm absolute decrease in height. All women received 400 IU vitamin D and 500 mg calcium supplementation per day.
Quarterly IV Injection
The effectiveness and safety of BONIVA Injection 3 mg once every 3 months were demonstrated in a randomized, double-blind, multinational, noninferiority study (DIVA Study) in 1358 women with postmenopausal osteoporosis (L2-L4 lumbar spine BMD, T score below -2.5 SD at baseline). The control group received BONIVA 2.5 mg daily oral tablets. The primary efficacy parameter was the relative change from baseline to 1 year of treatment in lumbar spine BMD, which was compared between the intravenous injection and the daily oral treatment groups. All patients received 400 IU vitamin D and 500 mg calcium supplementation per day.
Effect on Vertebral Fracture
BONIVA 2.5 mg daily oral tablet significantly reduced the incidence of new vertebral and of new and worsening vertebral fractures (Daily Oral Tablet – Treatment Study). Over the course of the 3-year study, the risk for vertebral fracture was 9.6% in the placebo-treated women and 4.7% in the women treated with BONIVA 2.5 mg daily oral tablet (p<0.001) (see Table 1). In an unapproved regimen, intermittent oral administration of 20 mg BONIVA, involving a 9- to 10-week drug-free interval, produced a statistically significant reduction (50%) in the incidence of new vertebral fractures, similar to that seen with the daily oral 2.5 mg regimen.
| Proportion of Patients with Fracture (%) | ||||
|---|---|---|---|---|
| Placebo n=975 | BONIVA 2.5 mg Daily n=977 | Absolute Risk Reduction (%) 95% CI | Relative Risk Reduction (%) 95% CI |
|
| New Vertebral Fracture | 9.6 | 4.7 | 4.9 | 52 † |
| 0-3 Year | (2.3, 7.4) | (29, 68) | ||
| New and Worsening Vertebral Fracture | 10.4 | 5.1 | 5.3 | 52 |
| 0-3 Year | (2.6, 7.9) | (30, 67) | ||
| Clinical (Symptomatic) Vertebral Fracture | 5.3 | 2.8 | 2.5 | 49 |
| 0-3 Year | (0.6, 4.5) | (14, 69) | ||
Effect on Nonvertebral Fractures
There was a similar number of nonvertebral osteoporotic fractures at 3 years reported in women treated with BONIVA 2.5 mg daily oral tablet [9.1%, (95% CI: 7.1%, 11.1%)] and placebo [8.2%, (95% CI: 6.3%, 10.2%)]. The two treatment groups were also similar with regard to the number of fractures reported at the individual non-vertebral sites: pelvis, femur, wrist, forearm, rib, and hip (Daily Oral Tablet - Treatment Study).
Effect on Bone Mineral Density (BMD)
Daily Oral Tablet - Treatment Study: BONIVA 2.5 mg daily oral tablet significantly increased BMD at the lumbar spine and hip relative to treatment with placebo. In the 3-year osteoporosis treatment study, BONIVA 2.5 mg daily oral tablet produced increases in lumbar spine BMD that were progressive over 3 years of treatment and were statistically significant relative to placebo at 6 months and at all later time points. Lumbar spine BMD increased by 6.4% after 3 years of treatment with BONIVA 2.5 mg daily oral tablet compared with 1.4% in the placebo group. Table 2 displays the significant increases in BMD seen at the lumbar spine, total hip, femoral neck, and trochanter compared to placebo. Thus, overall BONIVA 2.5 mg daily oral tablet reverses the loss of BMD, a central factor in the progression of osteoporosis.
| Placebo | BONIVA 2.5 mg | |
|---|---|---|
|
||
| Lumbar Spine | 1.4 (n=693) | 6.4 (n=712) |
| Total Hip | -0.7 (n=638) | 3.1 (n=654) |
| Femoral Neck | -0.7 (n=683) | 2.6 (n=699) |
| Trochanter | 0.2 (n=683) | 5.3 (n=699) |
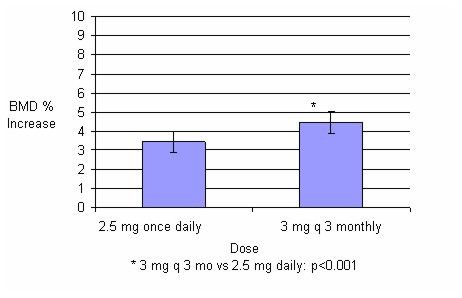
Quarterly IV Injection– DIVA Study: In the ITT efficacy analysis, the least-squares
mean increase at 1 year in lumbar spine BMD in patients (n=429) treated
with BONIVA Injection 3 mg once every 3 months (4.5%) was statistically
superior to that in patients (n=434) treated with daily oral tablets
(3.5%). The mean difference between groups was 1.05% (95% CI: 0.53%,
1.57%; p<0.001; see Figure 1). The mean increases
from baseline in total hip BMD at 1 year were 2.1% in the BONIVA Injection
3 mg once every 3 months group and 1.5% in the BONIVA 2.5 mg daily
oral tablet group. Consistently higher BMD increases at the femoral
neck and trochanter were also observed following BONIVA Injection
3 mg once every 3 months compared to BONIVA 2.5 mg daily oral tablet.
Figure 1 Mean Percent Change (95% CI) from Baseline in Lumbar Spine BMD at One Year in Patients Treated with BONIVA 2.5 mg Daily Oral Tablet or BONIVA Injection 3 mg Once Every 3 Months
Bone Histology
The effects of BONIVA 2.5 mg daily oral tablet on bone histology were evaluated in iliac crest biopsies from 16 women after 22 months of treatment and 20 women after 34 months of treatment. The histological analysis of bone biopsies showed bone of normal quality and no indication of osteomalacia or a mineralization defect.
The histological analysis of bone biopsies after 22 months of treatment with 3 mg intravenous ibandronate every 3 months (n=30) or 23 months of treatment with 2 mg intravenous ibandronate every 2 months (n=27) in women with postmenopausal osteoporosis showed bone of normal quality and no indication of a mineralization defect.
Animal Pharmacology
Animal studies have shown that ibandronate is an inhibitor of osteoclast-mediated bone resorption. In the Schenk assay in growing rats, ibandronate inhibited bone resorption and increased bone volume, based on histologic examination of the tibial metaphyses. There was no evidence of impaired mineralization at the highest dose of 5 mg/kg/day (subcutaneously), which is 1000 times the lowest antiresorptive dose of 0.005 mg/kg/day in this model, and 5000 times the optimal antiresorptive dose of 0.001 mg/kg/day in the aged ovariectomized rat. This indicates that BONIVA Injection administered at a therapeutic dose is unlikely to induce osteomalacia.
Long-term daily or intermittent administration of ibandronate to ovariectomized rats or monkeys was associated with suppression of bone turnover and increases in bone mass. Vertebral BMD, trabecular density, and biomechanical strength were increased dose-dependently in rats and monkeys, at doses up to 8 to 4 times the human intravenous dose of 3 mg every 3 months, based on cumulative dose normalized for body surface area (mg/m2) and AUC comparison, respectively. Ibandronate maintained the positive correlation between bone mass and strength at the ulna and femoral neck. New bone formed in the presence of ibandronate had normal histologic structure and did not show mineralization defects.
INDICATIONS AND USAGE
BONIVA Injection is indicated for the treatment of osteoporosis in postmenopausal women.
In postmenopausal women with osteoporosis, BONIVA increases BMD and reduces the incidence of vertebral fractures (see CLINICAL PHARMACOLOGY: Clinical Studies). Osteoporosis may be confirmed by the presence or history of osteoporotic fracture or by a finding of low bone mass (BMD more than 2.0 standard deviations below the premenopausal mean [ie, T-score]).
CONTRAINDICATIONS
- Known hypersensitivity to BONIVA Injection or to any of its excipients
- Uncorrected hypocalcemia (see PRECAUTIONS: General)
WARNINGS
BONIVA Injection, like other bisphosphonates administered intravenously, may cause a transient decrease in serum calcium values (see PRECAUTIONS).
BONIVA Injection must only be administered intravenously. Care must be taken not to administer BONIVA Injection intra-arterially or paravenously as this could lead to tissue damage.
Do not administer BONIVA Injection by any other route of administration. The safety and efficacy of BONIVA Injection following non-intravenous routes of administration have not been established.
PRECAUTIONS
General
Mineral Metabolism
Hypocalcemia, hypovitaminosis D, and other disturbances of bone and mineral metabolism must be effectively treated before starting BONIVA Injection therapy. Adequate intake of calcium and vitamin D is important in all patients. Patients must receive supplemental calcium and vitamin D.
Renal Impairment
Treatment with intravenous bisphosphonates has been associated with renal toxicity manifested as deterioration in renal function (ie, increased serum creatinine) and in rare cases, acute renal failure. No cases of acute renal failure were observed in controlled clinical trials in which intravenous BONIVA was administered as a 15- to 30-second bolus. The risk of serious renal toxicity with other intravenous bisphosphonates appears to be inversely related to the rate of drug administration.
Patients who receive BONIVA Injection should have serum creatinine measured prior to each dosage administration. Patients with concomitant diseases that have the potential for adverse effects on the kidney or patients who are taking concomitant medications that have the potential for adverse effects on the kidney should be assessed, as clinically appropriate. Treatment should be withheld for renal deterioration.
BONIVA Injection should not be administered to patients with severe renal impairment (ie, patients with serum creatinine >200µmol/L [2.3 mg/dL] or creatinine clearance [measured or estimated]<30 mL/min).
Jaw Osteonecrosis
Osteonecrosis, primarily in the jaw, has been reported in patients treated with bisphosphonates. Most cases have been in cancer patients undergoing dental procedures, but some have occurred in patients with postmenopausal osteoporosis or other diagnoses. Known risk factors for osteonecrosis include a diagnosis of cancer, concomitant therapies (eg, chemotherapy, radiotherapy, corticosteroids), and co-morbid disorders (eg, anemia, coagulopathy, infection, pre-existing dental disease). Most reported cases have been in patients treated with bisphosphonates intravenously but some have been in patients treated orally.
For patients who develop osteonecrosis of the jaw (ONJ) while on bisphosphonate therapy, dental surgery may exacerbate the condition. For patients requiring dental procedures, there are no data available to suggest whether discontinuation of bisphosphonate treatment reduces the risk of ONJ. Clinical judgment of the treating physician should guide the management plan of each patient based on individual benefit/risk assessment.
Musculoskeletal Pain
In postmarketing experience, severe and occasionally incapacitating bone, joint, and/or muscle pain has been reported in patients taking bisphosphonates that are approved for the prevention and treatment of osteoporosis (see ADVERSE REACTIONS). However, such reports have been infrequent. This category of drugs includes BONIVA (ibandronate sodium) Injection. Most of the patients were postmenopausal women. The time to onset of symptoms varied from one day to several months after starting the drug. Most patients had relief of symptoms after stopping. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate.
Information for Patients
BONIVA Injection must be administered intravenously only by a health care professional. Patients should be instructed to read the Patient Information Leaflet carefully before BONIVA Injection is administered and to re-read it each time the prescription is renewed.
BONIVA Injection should be administered once every 3 months. If the dose is missed, the injection should be administered as soon as it can be rescheduled. Thereafter, injections should be scheduled every 3 months from the date of the last injection. Do not administer BONIVA Injection more frequently than once every 3 months.
Patients must receive supplemental calcium and vitamin D.
Drug Interactions
See CLINICAL PHARMACOLOGY: Drug Interactions
Drug/Laboratory Test Interactions
Bisphosphonates are known to interfere with the use of bone-imaging agents. Specific studies with ibandronate have not been performed.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 104-week carcinogenicity study, doses of 3, 7, or 15 mg/kg/day were administered by oral gavage to Wistar rats (systemic exposures in males and females up to 3 and 1 times, respectively, human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison). There were no significant drug-related tumor findings in male or female rats. In a 78-week carcinogenicity study, doses of 5, 20, or 40 mg/kg/day were administered by oral gavage to NMRI mice (exposures in males and females up to 96 and 14 times, respectively, human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison). There were no significant drug-related tumor findings in male or female mice. In a 90-week carcinogenicity study, doses of 5, 20, or 80 mg/kg/day were administered in the drinking water to NMRI mice. A dose-related increased incidence of adrenal subcapsular adenoma/carcinoma was observed in female mice, which was statistically significant at 80 mg/kg/day (32 to 51 times human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison). The relevance of these findings to humans is unknown.
Mutagenesis
There was no evidence for a mutagenic or clastogenic potential of ibandronate in the following assays: in vitro bacterial mutagenesis assay in Salmonella typhimurium and Escherichia coli (Ames test), mammalian cell mutagenesis assay in Chinese hamster V79 cells, and chromosomal aberration test in human peripheral lymphocytes, each with and without metabolic activation. Ibandronate was not genotoxic in the in vivo mouse micronucleus tests for chromosomal damage.
Impairment of Fertility
In female rats treated from 14 days prior to mating through gestation, decreases in fertility, corpora lutea and implantation sites, and increased preimplantation loss were observed at an intravenous dose of 1.2 mg/kg/day (117 times human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison). In male rats treated for 28 days prior to mating, a decrease in sperm production and altered sperm morphology were observed at intravenous doses ≥0.3 mg/kg/day (≥40 times human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison).
Pregnancy
Pregnancy Category C
In pregnant rats given intravenous doses of 0.05, 0.15, or 0.5 mg/kg/day from Day 17 post-coitum until Day 20 post-partum, ibandronate treatment resulted in dystocia, maternal mortality, and early postnatal pup loss in all dose groups (≥2 times human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison). Reduced body weight at birth was observed at 0.15 and 0.5 mg/kg/day (≥4 times human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison). Pups exhibited abnormal odontogeny that decreased food consumption and body weight gain at 0.15 and 0.5 mg/kg/day (≥18 times human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison). Periparturient mortality has also been observed with other bisphosphonates and appears to be a class effect related to inhibition of skeletal calcium mobilization resulting in hypocalcemia and dystocia.
Exposure of pregnant rats during the period of organogenesis resulted in an increased fetal incidence of RPU (renal pelvis ureter) syndrome at an intravenous dose of 1 mg/kg/day (≥47 times human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison). In this spontaneous delivery study, dystocia was counteracted by perinatal calcium supplementation. In rat studies with intravenous dosing during gestation, fetal weight and pup growth were reduced at doses ≥0.1 mg/kg/day (≥5 times human exposure at the recommended intravenous dose of 3 mg every 3 months, based on cumulative AUC comparison).
In pregnant rabbits given intravenous doses of 0.03, 0.07 or 0.2 mg/kg/day during the period of organogenesis, maternal mortality, reduced maternal body weight gain, decreased litter size due to increased resorption rate, and decreased fetal weight were observed at 0.2 mg/kg/day (19 times the recommended human intravenous dose of 3 mg every 3 months, based on cumulative body surface area comparison, mg/m2).
Bisphosphonates are incorporated into the bone matrix, from where they are gradually released over periods of weeks to years. The extent of bisphosphonate incorporation into adult bone, and hence, the amount available for release back into the systemic circulation, is directly related to the total dose and duration of bisphosphonate use. Although there are no data on fetal risk in humans, bisphosphonates do cause fetal harm in animals, and animal data suggest that uptake of bisphosphonates into fetal bone is greater than into maternal bone. Therefore, there is a theoretical risk of fetal harm (eg, skeletal and other abnormalities) if a woman becomes pregnant after completing a course of bisphosphonate therapy. The impact of variables such as time between cessation of bisphosphonate therapy to conception, the particular bisphosphonate used, and the route of administration (intravenous versus oral) on this risk has not been established.
There are no adequate and well-controlled studies in pregnant women. BONIVA Injection should be used during pregnancy only if the potential benefit justifies the potential risk to the mother and fetus.
Nursing Mothers
In lactating rats treated with intravenous doses of 0.08 mg/kg, ibandronate was present in breast milk at concentrations of 8.1 to 0.4 ng/mL from 2 to 24 hours after dose administration. Concentrations in milk averaged 1.5 times plasma concentrations. It is not known whether BONIVA is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when BONIVA Injection is administered to a nursing woman.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Of the patients receiving BONIVA Injection 3 mg every 3 months for 1 year (DIVA study), 51% were over 65 years of age. No overall differences in effectiveness or safety were observed between these patients and younger patients, but greater sensitivity in some older individuals cannot be ruled out.
ADVERSE REACTIONS
Daily Oral Tablet
Treatment with BONIVA 2.5 mg daily oral tablet was studied in over 3900 patients in postmenopausal osteoporosis trials of up to 3 years duration. The overall adverse event profile of BONIVA 2.5 mg once daily tablet in these studies was similar to that of placebo.
Most adverse events were mild or moderate and did not lead to discontinuation. The incidence of serious adverse events was 20% in the placebo group and 23% in the BONIVA 2.5 mg daily oral tablet group. The percentage of patients who withdrew from treatment due to adverse events was approximately 17% in both the BONIVA 2.5 mg daily oral tablet group and the placebo group. Overall, and according to body system, there was no difference between BONIVA daily oral tablet and placebo, with adverse events of the digestive system being the most common reason for withdrawal.
Table 3 lists adverse events from the Treatment and Prevention Studies reported in ≥2% of patients and in more patients treated with BONIVA 2.5 mg daily oral tablet than patients treated with placebo. Adverse events are shown without attribution of causality.
| Body System | Placebo % (n=1134) | BONIVA 2.5 mg daily % (n=1140) |
|---|---|---|
| Body as a Whole | ||
| Back Pain | 12.2 | 13.5 |
| Pain in Extremity | 6.4 | 7.8 |
| Infection | 3.4 | 4.3 |
| Asthenia | 2.3 | 3.5 |
| Allergic Reaction | 1.9 | 2.5 |
| Digestive System | ||
| Dyspepsia | 9.8 | 11.9 |
| Diarrhea | 5.0 | 6.8 |
| Tooth Disorder | 2.3 | 3.5 |
| Vomiting | 2.1 | 2.7 |
| Gastritis | 1.9 | 2.2 |
| Metabolic and Nutritional Disorders | ||
| Hypercholesterolemia | 4.2 | 4.8 |
| Musculoskeletal System | ||
| Myalgia | 5.1 | 5.7 |
| Joint Disorder | 3.3 | 3.6 |
| Arthritis | 2.7 | 3.2 |
| Nervous System | ||
| Headache | 5.8 | 6.5 |
| Dizziness | 2.6 | 3.7 |
| Vertigo | 2.5 | 3.0 |
| Nerve Root Lesion | 1.9 | 2.2 |
| Respiratory System | ||
| Upper Respiratory Infection | 33.2 | 33.7 |
| Bronchitis | 6.8 | 10.0 |
| Pneumonia | 4.3 | 5.9 |
| Pharyngitis | 1.5 | 2.5 |
| Urogenital System | ||
| Urinary Tract Infection | 4.2 | 5.5 |
Quarterly IV Injection – DIVA Study
In a 1-year, double-blind, multicenter study comparing BONIVA Injection administered intravenously as 3 mg every 3 months to BONIVA 2.5 mg daily oral tablet in women with postmenopausal osteoporosis, the overall safety and tolerability profiles of the two dosing regimens were similar. The incidence of serious adverse events was 8.0% in the BONIVA 2.5 mg daily group and 7.5% in the BONIVA Injection 3 mg once every 3 months group. The percentage of patients who withdrew from treatment due to adverse events was approximately 6.7% in the BONIVA 2.5 mg daily group and 8.5% in the BONIVA Injection 3 mg every 3 months group.
Table 4 lists the adverse events reported in >2% of patients without attribution of causality.
| Body System/Adverse Event | BONIVA 2.5 mg Daily (Oral) % (n=465) | BONIVA 3 mg q 3 mo (IV) % (n=469) |
|---|---|---|
|
||
| Infections and Infestations | ||
| Influenza | 8.0 | 4.7 |
| Nasopharyngitis | 6.0 | 3.4 |
| Cystitis | 3.4 | 1.9 |
| Gastroenteritis | 3.4 | 1.5 |
| Urinary Tract Infection | 3.2 | 2.6 |
| Bronchitis | 2.8 | 2.1 |
| Upper Respiratory Tract Infection | 2.8 | 1.1 |
| Gastrointestinal Disorders | ||
| Abdominal Pain* | 5.6 | 5.1 |
| Dyspepsia | 4.3 | 3.6 |
| Nausea | 4.3 | 2.1 |
| Constipation | 4.1 | 3.4 |
| Diarrhea | 2.4 | 2.8 |
| Gastritis | 2.2 | 1.9 |
| Musculoskeletal and Connective Tissue Disorders | ||
| Arthralgia | 8.6 | 9.6 |
| Back Pain | 7.5 | 7.0 |
| Localized Osteoarthritis | 2.4 | 1.5 |
| Pain in Extremity | 2.2 | 2.8 |
| Myalgia | 0.9 | 2.8 |
| Nervous System Disorders | ||
| Dizziness | 2.8 | 1.9 |
| Headache | 2.6 | 3.6 |
| Vascular Disorders | ||
| Hypertension | 7.1 | 5.3 |
| Psychiatric Disorders | ||
| Insomnia | 2.6 | 1.1 |
| Depression | 2.2 | 1.3 |
| General Disorders and Administration Site Conditions | ||
| Influenza-like Illness† | 1.1 | 4.9 |
| Fatigue | 1.1 | 2.8 |
| Skin and Subcutaneous Tissue Disorders | ||
| Rash‡ | 2.8 | 2.3 |
| Metabolism and Nutrition Disorders | ||
| Hypercholesterolemia | 4.3 | 1.5 |
Acute Phase Reaction-like Events
Symptoms consistent with acute phase reaction (APR) have been reported with intravenous bisphosphonate use. The overall incidence of patients with APR-like events was higher in the intravenous treatment group (4% in the BONIVA 2.5 mg daily oral tablet group vs. 10% in the BONIVA Injection 3 mg once every 3 months group). These incidence rates are based on reporting of any of 33 potential APR-like symptoms within 3 days of an IV dose and for a duration of 7 days or less. In most cases, no specific treatment was required and the symptoms subsided within 24 to 48 hours.
Injection Site Reactions
Local reactions at the injection site, such as redness or swelling, were observed infrequently, but at a higher incidence in patients treated with BONIVA Injection 3 mg every 3 months (<2%; 8/469) than in patients treated with placebo injections (<1%; 1/465). In most cases, the reaction was of mild to moderate severity.
Ocular Adverse Events
Bisphosphonates may be associated with ocular inflammation such as uveitis and scleritis. In some cases, these events did not resolve until the bisphosphonate was discontinued.
Laboratory Test Findings
There were no clinically significant changes from baseline values or shifts in any laboratory variable with oral ibandronate. As expected with bisphosphonate treatment, a decrease in total alkaline phosphatase levels was seen with 2.5 mg daily oral ibandronate compared to placebo. There was no difference compared with placebo for laboratory abnormalities indicative of hepatic or renal dysfunction, hypocalcemia, or hypophosphatemia. There also was no evidence that BONIVA Injection 3 mg every 3 months induced clinically significant laboratory abnormalities indicative of hepatic or renal dysfunction compared to BONIVA 2.5 mg daily oral tablet.
OVERDOSAGE
No cases of overdose were reported in premarketing studies with BONIVA Injection. Intravenous overdosage may result in hypocalcemia, hypophosphatemia, and hypomagnesemia. Clinically relevant reductions in serum levels of calcium, phosphorus, and magnesium should be corrected by intravenous administration of calcium gluconate, potassium or sodium phosphate, and magnesium sulfate, respectively.
Dialysis would not be beneficial unless it is administered within 2 hours following the overdose.
DOSAGE AND ADMINISTRATION
The recommended dose of BONIVA Injection for the treatment of postmenopausal osteoporosis is 3 mg every 3 months (see INDICATIONS AND USAGE) administered over a period of 15 to 30 seconds.
No cases of acute renal failure were observed in controlled clinical trials in which intravenous BONIVA was administered as a 15- to 30-second bolus. The risk of serious renal toxicity with other intravenous bisphosphonates appears to be inversely related to the rate of drug administration (see PRECAUTIONS).
BONIVA Injection must be administered by a health care professional.
BONIVA Injection must only be administered intravenously (see WARNINGS). Care must be taken not to administer BONIVA Injection intra-arterially or paravenously as this could lead to tissue damage.
Do not administer BONIVA Injection by any other route of administration. The safety and efficacy of BONIVA Injection following non-intravenous routes of administration have not been established.
Administer BONIVA Injection using the enclosed needle. Prefilled syringes are for single use only. Discard unused portion.
BONIVA Injection must not be mixed with calcium-containing solutions or other intravenously administered drugs.
Parenteral drug products should be inspected visually for particulate matter and discoloration before administration, and not used if particulate matter is visible or product is discolored. Prefilled syringes with particulate matter or discoloration should not be used.
If the dose is missed, BONIVA Injection should be administered as soon as it can be rescheduled. Thereafter, injections should be scheduled every 3 months from the date of the last injection. Do not administer BONIVA Injection (3 mg) more frequently than once every 3 months.
Patients must receive supplemental calcium and vitamin D (see PRECAUTIONS: Information for Patients).
Patients with Hepatic Impairment
No dose adjustment is necessary (see CLINICAL PHARMACOLOGY: Special Populations).
Patients with Renal Impairment
No dose adjustment is necessary for patients with mild or moderate renal impairment where creatinine clearance is equal to or greater than 30 mL/min.
BONIVA Injection should not be administered to patients with severe renal impairment, ie, patients with serum creatinine >200 µmol/L (2.3 mg/dL) or creatinine clearance (measured or estimated) <30 mL/min (see CLINICAL PHARMACOLOGY: Special Populations).
Geriatric Patients
No dosage adjustment is necessary in the elderly (see PRECAUTIONS: Geriatric Use).
HOW SUPPLIED
One prefilled syringe of BONIVA Injection (ibandronate sodium), 3 mg/3 mL single-use, clear glass prefilled syringe, in a box with 1 needle and 2 alcohol swabs (NDC 0004-0188-09).
Each syringe is a 5 mL (5 cc) volume syringe supplied with a 23-gauge, 3/4 inch needle with wings, needle-stick protection device, and 3-inch plastic tubing for attachment.
Storage
Store at 25°C (77°F); excursions permitted between 15° and 30°C (59° and 86°F) [see USP Controlled Room Temperature].
BONIVA is a registered trademark of Roche Therapeutics Inc.
Distributed by:
Roche
Pharmaceuticals
Roche Laboratories Inc.
340 Kingsland Street
Nutley, New Jersey 07110–1199
Co-promoted by Roche Laboratories Inc. and
GlaxoSmithKline
Research Triangle Park, NC 27709
February 2007
Copyright © 2007 by Roche Laboratories Inc. All rights reserved.
| BONIVA
ibandronate sodium injection, solution |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021858 | 01/06/2006 | |
| Labeler - Hoffmann-La Roche Inc (002191211) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Vetter Pharma-Fertigung GmbH & Co KG | 344217323 | Analysis, Manufacture | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Roche Diagnostics GmbH | 315028860 | Analysis, Manufacture | |