ALPHANATE
-
antihemophilic factor/von willebrand factor complex (human)
Grifols Biologicals Inc.
----------
|
|||||||||||||||||||||||
FULL PRESCRIBING INFORMATION
1. INDICATIONS AND USAGE
1.1 Hemophilia A or Acquired Factor VIII Deficiency
Antihemophilic Factor/von Willebrand Factor Complex (Human), Alphanate®, is indicated for the prevention and control of bleeding in patients with Factor VIII deficiency due to hemophilia A or acquired Factor VIII deficiency.1
1.2 von Willebrand Disease
Antihemophilic Factor/von Willebrand Factor Complex (Human), Alphanate®, is also indicated for surgical and/or invasive procedures in patients with von Willebrand Disease (VWD) in whom desmopressin (DDAVP®) is either ineffective or contraindicated. It is not indicated for patients with severe VWD (Type 3) undergoing major surgery.
2. DOSAGE AND ADMINISTRATION
Following reconstitution with the supplied diluent, Alphanate® should be administered intravenously within three hours after reconstitution to avoid the potential ill effect of any inadvertent bacterial contamination occurring during reconstitution. Alphanate® is administered by injection (plastic disposable syringes are recommended). Administer at room temperature, do not refrigerate after reconstitution, and discard any unused contents into the appropriate safety container.
Antihemophilic Factor (AHF) potency (Factor VIII:C activity) is expressed in International Units (IU) on the product label. Additionally, each vial of Alphanate® also contains von Willebrand Factor:Ristocetin Cofactor (VWF:RCof) activity in IU for the treatment of VWD.
2.1 Hemophilia A
Dosing requirements and frequency of dosing is calculated on the basis of an expected initial response of 2% of normal FVIII:C increase per FVIII:C IU/kg body weight administered.2,3 The in vivo increase in plasma Factor VIII can therefore be estimated by multiplying the dose of AHF per kilogram of body weight (FVIII:C IU/kg) by 2%. Thus, an administered AHF dose of 50 IU/kg will be expected to increase the circulating Factor VIII level by 100% of normal (100 IU/dL). The following formulas and examples illustrate these principles:
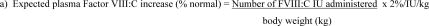
Example: A 70 kg adult administered AHF 2100 IU:

Example: A 15 kg child with a baseline plasma FVIII level of 0%. To increase the plasma Factor VIII concentration to 100% of normal, the dosage required is as follows:
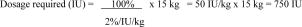
The following dosages are presented as general guidance. It should be emphasized that the dosage of Alphanate® required for hemostasis must be individualized according to the needs of the patient, the severity of the deficiency, the severity of the hemorrhage, the presence of inhibitors, and the FVIII level desired. Adequacy of treatment must be judged by the clinical effects and situation and thus, the dosage may vary with individual cases.
| Hemorrhagic event | Dosage (AHF FVIII:C IU/kg Body Weight) |
Minor hemorrhage:
| FVIII:C levels should be brought to 30% of normal (15 FVIII IU/kg twice daily) until hemorrhage stops and healing has been achieved (1–2 days). |
Moderate hemorrhage:
| FVIII:C levels should be brought to 50% (25 FVIII IU/kg twice daily). Treatment should continue until healing has been achieved (2–7 days, on average). |
Major hemorrhage:
| FVIII:C levels should be brought to 80–100% for at least 3–5 days (40–50 FVIII IU/kg twice daily). Following this treatment period, FVIII levels should be maintained at 50% (25 FVIII IU/kg twice daily) until healing has been achieved. Major hemorrhages may require treatment for up to 10 days. |
| Surgery | Prior to surgery, the levels of FVIII:C should be brought to 80–100% of normal (40–50 FVIII IU/kg). For the next 7–10 days, or until healing has been achieved, the patient should be maintained at 60–100% FVIII levels (25–50 FVIII IU/kg twice daily). |
Dosing requirements and frequency of dosing is calculated on the basis of an expected initial response of 2% FVIII:C increase per FVIII:C IU/kg body weight (i.e., 2% per IU/kg) and an average half-life for FVIII:C of 12 hours.4,5 If dosing studies have determined that a particular patient exhibits a lower than expected response, the dose should be adjusted accordingly. Failure to achieve the expected plasma FVIII:C level or to control bleeding after an appropriately calculated dosage may be indicative of the development of an inhibitor (an antibody to FVIII:C). Its presence should be documented and the inhibitor level quantitated by appropriate laboratory procedures. Treatment with AHF in such cases must be individualized .6-8
Plasma factor VIII levels should be monitored periodically to evaluate individual patient response to the dosage regime.
2.2 von Willebrand Disease
The following table provides dosing guidelines for pediatric and adult patients with von Willebrand Disease. 9-12
The amount of VWF:RCof and Factor VIII contained in each vial of Alphanate® is indicated on the vial's label. The ratio of VWF:RCof to Factor VIII in Alphanate® varies by lot, so dosage should be re-evaluated whenever lot selection is changed.
| Bleeding Prophylaxis for Surgical or Invasive Procedures | Dosage (AHF VWF:RCof IU/kg Body Weight) |
| Adult | Pre-operative dosage: 60 VWF:RCof IU/kg body weight. Subsequent infusions: 40 to 60 VWF:RCof IU/kg body weight at 8 to 12 hour intervals as clinically needed. Dosing may be reduced after the third postoperative day. Continue treatment until healing is complete. |
| Minor procedure: VWF activity of 40%-50% during 1 to 3 days postoperative. | |
| Major procedure: VWF activity of 40%-50% during at least 3 to 7 days postoperative. | |
| Pediatric | Initial dosage: 75 VWF:RCof IU/kg body weight. Subsequent infusions: 50 to 75 VWF:RCof IU/kg body weight at 8 to 12 hour intervals as clinically needed. Dosing may be reduced after the third postoperative day. Continue treatment until healing is complete. |
2.3 Reconstitution
Always Use Aseptic Technique
- Warm diluent (Sterile Water for Injection, USP) and concentrate (Alphanate®) to at least room temperature (but not above 37 °C).
- Remove plastic caps from the diluent and concentrate vials.
- Swab the exposed stopper surfaces with a cleansing agent such as alcohol. Do not leave excess cleansing agent on the stoppers.
- Remove cover from one end of the double-ended transfer needle. Insert the exposed end of the needle through the center of the stopper in the DILUENT vial.
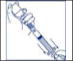
- Remove plastic cap from the other end of the double-ended transfer needle now seated in the stopper of the diluent vial. To reduce any foaming, invert the vial of diluent and insert the exposed end of the needle through the center of the stopper in the CONCENTRATE vial at an angle, making certain that the diluent vial is always above the concentrate vial. The angle of insertion directs the flow of diluent against the side of the concentrate vial. Refer to Figure 1. There should be enough vacuum in the vial to transfer all of the diluent.
- Disconnect the two vials by removing the transfer needle from the diluent vial stopper. Remove the double-ended transfer needle from the concentrate vial and discard the needle into the appropriate safety container.
- Let the vial stand until contents are in solution, then GENTLY swirl until all concentrate is dissolved. Reconstitution requires less than 5 minutes.
- DO NOT SHAKE THE CONTENTS OF THE VIAL. DO NOT INVERT THE CONCENTRATE VIAL UNTIL READY TO WITHDRAW CONTENTS.
- Use as soon as possible after reconstitution.
- After reconstitution, parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. When reconstitution procedure is strictly followed, a few small particles may occasionally remain. The microaggregate filter will remove particles and the labeled potency will not be reduced.
2.4 Administration by Syringe
Use Aseptic Technique
- Peel cover from microaggregate filter package and securely install the syringe into the exposed Luer inlet of the filter, using a slight clockwise twisting motion.
- Remove filter from packaging. Remove protective cover from the spike end of the filter.
- Pull back plunger drawing sufficient air into the syringe to allow reconstituted product to be withdrawn as described in the next step.
- Insert the spike end of the filter into the reconstituted concentrate vial. Inject air (Figure 2a) and withdraw the reconstituted product from the vial into the syringe (Figure 2b).
- Remove the filter from the syringe; discard the filter and the empty concentrate vial, into the appropriate safety container. Attach syringe to an infusion set, expel air from the syringe and infusion set. Perform venipuncture and administer slowly at a rate not exceeding 10 mL/minute.
- If the patient is to receive more than one vial of concentrate, the infusion set will allow administration of multiple vials to be performed with a single venipuncture.
- Discard all administration equipment after use into the appropriate safety container. Do not reuse.
3. DOSAGE FORM AND STRENGTHS
Alphanate® is a sterile, lyophilized powder for injection. It is available in the following potencies:
- 250 IU/5 mL single dose vial
- 500 IU/5 mL single dose vial
- 1000 IU/10 mL single dose vial
- 1500 IU/10 mL single dose vial
4. CONTRAINDICATIONS
None.
5. WARNINGS AND PRECAUTIONS
5.1 Thromboembolic Events
Thromboembolic events have been reported in von Willebrand Disease patients receiving Antihemophilic Factor/von Willebrand Factor Complex replacement therapy, especially in the setting of known risk factors for thrombosis.13,14 Early reports might indicate a higher incidence in females. In addition, endogenous high levels of FVIII have also been associated with thrombosis but no causal relationship has been established. In all VWD patients in situations of high thrombotic risk receiving coagulation factor replacement therapy, caution should be exercised and antithrombotic measures should be considered. See also ADVERSE REACTIONS (6.1) and PATIENT COUNSELING INFORMATION (17.1).
5.2 Infections
Because Antihemophilic Factor/von Willebrand Factor Complex (Human), Alphanate® is made from pooled human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, and theoretically, the Creutzfeldt-Jakob disease (CJD) agent. Stringent procedures designed to reduce the risk of adventitious agent transmission have been employed in the manufacture of this product, from the screening of plasma donors and the collection and testing of plasma, through the application of viral elimination/reduction steps such as solvent detergent and heat treatment in the manufacturing process. Despite these measures, such products can still potentially transmit disease; therefore, the risk of infectious agents cannot be totally eliminated. All infections thought by a physician possibly to have been transmitted by this product should be reported to the manufacturer at 888-GRIFOLS (888-474-3657) for USA and 323-225-2221 for international. The physician should weigh the risks and benefits of the use of this product and should discuss these with the patient. See also PATIENT COUNSELING INFORMATION (17.2).
Individuals who receive infusions of blood or plasma products may develop signs and/or symptoms of some viral infections, particularly hepatitis C.15,16 Incubation in a solvent detergent mixture during the manufacturing process is designed to reduce the risk of transmitting viral infection.15,16 However, medical opinion encourages hepatitis A and hepatitis B vaccinations for patients with hemophilia at birth or at the time of diagnosis.
Nursing personnel, and others who administer this material, should exercise appropriate caution when handling due to the risk of exposure to viral infection.
5.3 Inhibitor Formation
Rapid administration of a Factor VIII concentrate may result in vasomotor reactions. Alphanate® should not be administered at a rate exceeding 10 mL/minute.
Some patients develop inhibitors to Factor VIII. These inhibitors are circulating antibodies (i.e., globulins) that neutralize the procoagulant activity of Factor VIII. No studies have been conducted with Alphanate® to evaluate inhibitor formation. Therefore, it is not known whether there are greater, lesser or the same risks of developing inhibitors due to the use of this product than there are with other antihemophilic factor preparations. Patients with these inhibitors may not respond to treatment with Antihemophilic Factor/von Willebrand Factor Complex (Human), or the response may be much less than would otherwise be expected; therefore, larger doses of Antihemophilic Factor/von Willebrand Factor Complex (Human) are often required. The management of bleeding in patients with inhibitors requires careful monitoring, especially if surgical procedures are indicated.6-8 See also PATIENT COUNSELING INFORMATION (17.3).
Reports in the literature suggest that patients with Type 3, severe von Willebrand Disease, may occasionally develop alloantibodies to von Willebrand factor after replacement therapy.17 The risk of developing alloantibodies in patients with von Willebrand disease due to the use of this product is not known.
Unused contents should be discarded into the appropriate safety container. Administration equipment should be discarded after single use into the appropriate safety container. Components should not be re-sterilized.
5.4 Information for Patients
Patients should be informed of the early symptoms and signs of hypersensitivity reaction, including hives, generalized urticaria, chest tightness, dyspnea, wheezing, faintness, hypotension, and anaphylaxis. Patients should be advised to discontinue use of the product and contact their physician and/or seek immediate emergency care, depending on the severity of the reaction, if these symptoms occur.
Patients should be informed of a potential for viral infection such as parvovirus B19 or hepatitis A. Parvovirus B19 may most seriously affect seronegative pregnant women, or immunocompromised individuals. Patients should report any signs and symptoms of fever, sore throat, or joint soreness to the physician immediately.
6. ADVERSE REACTIONS
6.1 General
The most common adverse reactions may include urticaria, fever, chills, nausea, vomiting, headache, somnolence, or lethargy.
Occasionally, mild reactions occur following the administration of Antihemophilic Factor/von Willebrand Factor Complex (Human), such as allergic reactions, chills, nausea, or stinging at the infusion site.4 If a reaction is experienced, and the patient requires additional Antihemophilic Factor/von Willebrand Factor Complex (Human), product from a different lot should be administered.
Massive doses of Antihemophilic Factor/von Willebrand Factor Complex (Human) have rarely resulted in acute hemolytic anemia, increased bleeding tendency or hyperfibrinogenemia.5 Alphanate® contains blood group specific isoagglutinins and, when large and/or frequent doses are required in patients of blood groups A, B, or AB, the patient should be monitored for signs of intravascular hemolysis and falling hematocrit. Should this condition occur, thus leading to progressive hemolytic anemia, the administration of serologically compatible Type O red blood cells should be considered, the administration of Alphanate® should be discontinued, and alternative therapy should be considered.
Reports of thromboembolic events in VWD patients with other thrombotic risk factors receiving coagulation factor replacement therapy have been obtained from published literature. Early reports might indicate a higher incidence in females. Caution should be exercised and antithrombotic measures should be considered in all VWD patients in situations of high thrombotic risk. See WARNINGS AND PRECAUTIONS (5.1).
6.2 Adverse Reactions in VWD Patients from Clinical Studies
In clinical studies of Alphanate® (A-SD/HT) in patients with VWD, adverse reactions occurred in 6 of 38 (15.8%) subjects and 17 of 299 (5.7%) infusions. The most common adverse events were pruritus, pharyngitis (throat tightness), paresthesia and headache, edema of the face, rash and chills. Except for one instance of pruritus, which was considered moderate in severity, all the adverse events were assessed as mild in severity.
A single incident of pulmonary embolus was reported that was considered to have a possible relationship to the product. This subject received the dose of 60 VWF:RCof IU/kg body weight and the FVIII:C level achieved was 290%.
In the retrospective study, 3 out of 39 subjects (7.7%) experienced 6 adverse drug reactions. Four were considered mild and 2 were considered moderate; and no subject discontinued their treatment due to an adverse reaction. The adverse drug reactions were pruritus, paresthesia (2 events) and hemorrhage (all considered mild), and one event each of moderate hematocrit decrease and orthostatic hypotension.
Only one adverse event (pain) related to the treatment with heat-treated Alphanate® (A-SD/HT) was reported on the four pediatric patients with von Willebrand Disease during the course of the prospective study and none of the five subjects in the retrospective clinical study.18
6.3 Adverse Reaction Information from Spontaneous Reports
The following adverse reactions have been identified during post-approval use of Alphanate® (A-SD/HT). Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
These adverse reactions have been reported as swelling of the parotid gland, urticaria, nausea, shortness of breath, chest tightness, chills, fever, rigors, headache, flushing, vomiting, joint pain, seizure, pulmonary embolus, femoral venous thrombosis, itching and cardiorespiratory arrest.
7. DRUG INTERACTIONS
None known.
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C. Animal reproduction studies have not been conducted with Alphanate®. It is also not known whether Alphanate® can cause fetal harm when administered to a pregnant woman or affect reproductive capacity. Alphanate® should be given to a pregnant woman only if clearly needed.
8.4 Pediatric Use
8.4.1 Hemophilia A Indication
Clinical trials for safety and effectiveness in pediatric Hemophilia A patients 16 years of age and younger have not been conducted. During a well controlled half-life and recovery clinical trial in patients previously treated with Factor VIII concentrates for Hemophilia A, the single pediatric patient receiving Alphanate® (solvent detergent non-heat treated) responded similarly when compared with 12 adult patients.4 No adverse events were reported in either pediatric or adult patients with Alphanate®.
8.4.2 VWD Indication
Fifteen pediatric patients with von Willebrand Disease younger than 18 years of age were treated with non-heat (A-SD) and heat-treated (A-SD/HT) Alphanate® during the course of clinical studies.18 In the retrospective study, five patients younger than 18 years of age were treated with heat-treated (A-SD/HT) Alphanate®.
11. DESCRIPTION
Antihemophilic Factor/von Willebrand Factor Complex (Human), Alphanate® sterile, lyophilized concentrate of Factor VIII (AHF) and von Willebrand Factor (VWF), is intended for intravenous administration in the treatment of hemophilia A, acquired Factor VIII deficiency, and von Willebrand Disease (VWD).
Alphanate® is prepared from pooled human plasma by cryoprecipitation of Factor VIII, fractional solubilization, and further purification employing heparin-coupled, cross-linked agarose which has an affinity to the heparin binding domain of VWF/FVIII:C complex.19 The product is treated with a mixture of tri-n-butyl phosphate (TNBP) and polysorbate 80 to reduce the risks of transmission of viral infection. In order to provide an additional safeguard against potential non-lipid enveloped viral contaminants, the product is also subjected to an 80 °C heat treatment step for 72 hours. However, no procedure has been shown to be totally effective in removing viral infectivity from coagulation factor products.
Alphanate® is labeled with the antihemophilic factor potency (Factor VIII:C activity) in International Units (IU) per vial. Each vial of Alphanate® also contains the labeled amount of von Willebrand Factor:Ristocetin Cofactor (VWF:RCof) activity expressed in IU. An IU is defined by the current international standard established by the World Health Organization. One IU of Factor VIII or one IU of VWF:RCof is approximately equal to the amount of Factor VIII or VWF:RCof in 1 mL of freshly-pooled human plasma.
Alphanate® contains Albumin (Human) as a stabilizer, resulting in a final container concentrate with a specific activity of at least 5 FVIII:C IU/mg total protein. Prior to the addition of the Albumin (Human) stabilizer, the specific activity is significantly higher.
When reconstituted as directed, the composition of Alphanate® is as follows:
|
NMT = not more than |
|
|
NLT = not less than |
|
| Component | Concentration |
| Factor VIII:C activity | 40 – 180 IU/mL |
| VWF:RCof activity | NLT 0.4 VWF:RCof IU per 1 IU of FVIII:C (NLT 16 IU/mL) |
| Albumin (Human) | 0.3 – 0.9 g/100 mL |
| Calcium | NMT 5 mmol/L |
| Glycine | NMT 750 μg per FVIII:C IU |
| Heparin | NMT 1.0 U/mL |
| Histidine | 10 – 40 mmol/L |
| Imidazole | NMT 0.1 mg/mL |
| Arginine | 50 – 200 mmol/L |
| Polyethylene Glycol and Polysorbate 80 | NMT 1.0 μg per FVIII:C IU |
| Sodium | NLT 10 mEq/vial |
| Tri-n-butyl Phosphate (TNBP) | NMT 0.1 μg per FVIII:C IU |
Viral Reduction Capacity
The solvent detergent treatment process has been shown by Horowitz, et al., to provide a high level of viral inactivation without compromising protein structure and function.20 The susceptibility of human pathogenic viruses such as Human Immunodeficiency viruses (HIV), hepatitis viruses, as well as marker viruses such as Sindbis virus (SIN, a model for Hepatitis C virus) and Vesicular Stomatitis virus (VSV, a model for large, enveloped RNA virus), to inactivation by organic solvent detergent treatment has been discussed in the literature.21
In vitro inactivation studies to evaluate the solvent detergent treatment (0.3% Tri-n-butyl Phosphate and 1.0% Polysorbate 80) step in the manufacture of Alphanate® demonstrated a log inactivation of ≥ 11.1 for HIV-1, ≥ 6.1 for HIV-2, ≥ 4.1 for VSV and ≥ 4.7 for SIN. Since the number of virus particles inactivated by the process represents the maximum amount of virus added initially to the sample, these results indicate that all the virus added was killed to the assay limit of detection.18
Additional steps in the manufacturing process of Alphanate® were evaluated for virus elimination capability. The dry heat cycle of 80 °C for 72 hours was shown to inactivate greater than 5.8 logs of Hepatitis A virus (HAV).18 Precipitation with 3.5% polyethylene glycol (PEG) and heparin-actigel-ALD chromatography are additional steps studied using Bovine Herpes virus (BHV, a model for Hepatitis B virus), Bovine Viral Diarrhea virus (BVD, a second model for Hepatitis C virus), human Poliovirus Sabin type 2 (POL, a model for Hepatitis A virus), Canine Parvovirus (CPV, a model for Parvovirus B19) and HIV-1.
Table 3 summarizes the reduction factors for each virus validation study performed for the manufacturing process of Alphanate®.18 It must be stated that no treatment method has yet been shown capable of totally eliminating all potential infectious virus in preparations of coagulation factor concentrates.
|
Virus (Model Virus for) | 3.5% PEG
Precipitation | Solvent–Detergent | Column
Chromatography | Lyophilization | Dry Heat Cycle
(80 °C, 72 h) | Total Log
Removal |
| BHV (HBV) | < 1.0 | ≥ 8.0 | 7.6 | 1.3 | 2.1 | ≥ 19.0 |
| BVD (HCV) | < 1.0 | ≥ 4.5 | < 1.0 | < 1.0 | ≥ 4.9 | ≥ 9.4 |
| POL (HAV) | 3.3 | – | < 1.0 | 3.4 | ≥ 2.5 | ≥ 9.2 |
| CPV (B19) | 1.2 | – | < 1.0 | < 1.0 | 4.1 | 5.3 |
| VSV | – | ≥ 4.1 | – | – | – | ≥ 4.1 |
| SIN (HCV) | – | ≥ 4.7 | – | – | ≥ 4.7 | |
| HIV–1 | < 1.0 | ≥ 11.1 | ≥2.0 | – | – | ≥ 13.1 |
| HIV–2 | – | ≥ 6.1 | – | – | – | ≥ 6.1 |
| HAV | – | – | – | 2.1 | ≥ 5.8 | ≥ 7.9 |
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Antihemophilic Factor/von Willebrand Factor Complex (Human) (Factor VIII) and von Willebrand Factor (VWF) are constituents of normal plasma and are required for clotting. The administration of Alphanate® temporarily increases the plasma level of Factor VIII, thus minimizing the hazard of hemorrhage.22,23 Factor VIII is an essential cofactor in activation of Factor X leading to formation of thrombin and fibrin. VWF promotes platelet aggregation and platelet adhesion on damaged vascular endothelium; it also serves as a stabilizing carrier protein for the procoagulant protein Factor VIII.24,25
12.3 Pharmacokinetics
12.3.1 Pharmacokinetics in Hemophilia A
Following the administration of Alphanate® during clinical trials, the mean in vivo half-life of Factor VIII observed in 12 adult subjects with severe hemophilia A was 17.9 ± 9.6 hours. In this same study, the in vivo recovery was 96.7 ± 14.5% at 10 minutes postinfusion.18 Recovery at 10 minutes post-infusion was also determined as 2.4 ± 0.4 IU FVIII rise/dL plasma per IU FVIII infused/kg body weight.18
12.3.2 Pharmacokinetics in von Willebrand Disease (VWD)
A pharmacokinetic crossover study was conducted in 14 non-bleeding subjects with VWD (1 type 1, 2 type 2A, and 11 type 3) comparing the pharmacokinetics of Alphanate® SD/HT (A-SD/HT) and an earlier formulation, Alphanate® SD (A-SD), which was treated with solvent-detergent but was not heat-treated.18 Subjects received, in random order at least seven days apart, a single intravenous dose each of A-SD and A-SD/HT, 60 VWF:RCof IU/kg (75 VWF:RCof IU/kg in subjects younger than 18 years of age). Pharmacokinetic parameters were similar for the two preparations and indicated that they were biochemically equivalent. Pharmacokinetic analysis of A-SD/HT in the 14 subjects revealed the following results 18: the median plasma levels of VWF:RCof rose from 0.17 IU/dL [mean, 0.2 ± 0.08 IU/dL; range: 0.1 to 0.5 IU/dL] at baseline to 3.43 IU/dL [mean, 3.5 ± 1.47 IU/dL; range: 1.5 to 5.9 IU/dL] 15 minutes post-infusion; median plasma levels of FVIII:C rose from 0.08 IU/dL [mean, 0.2 ± 0.34 IU/dL; range: 0.0 to 1.2 IU/dL] to 2.14 IU/dL [mean, 2.4 ± 0.72 IU/dL; range: 1.4 to 3.9 IU/dL]. The median bleeding time (BT) prior to infusion was 30 minutes (mean, 28.8 ± 4.41 minutes; range: 13.5 to 30 minutes), which shortened to 10.38 minutes (mean, 10.4 ± 3.20 minutes; range: 6 to 16 minutes) 1 hour post-infusion.
Following infusion of A-SD/HT, the median half-lives for VWF:RCof, FVIII:C and VWF:Ag were 6.91 hours (mean, 7.46 ± 3.20 hours, range, 3.68 to 16.22 hours), 20.87 hours (mean, 21.52 ± 7.21 hours; range: 7.19 to 32.20 hours), and 12.66 hours (mean, 13.03 ± 2.12 hours: range: 10.34 to 17.45 hours), respectively. The median incremental in vivo recoveries of VWF:RCof and FVIII:C were 3.12 (IU/dL)/(IU/kg) [mean, 3.29 ± 1.46 (IU/dL)/(IU/kg); range: 1.3 to 5.7 (IU/dL)/(IU/kg)] for VWF:RCof and 1.94 (IU/dL)/(IU/kg) [mean, 2.14 ± 0.58 (IU/dL)/(IU/kg); range: 1.3 to 3.3 (IU/dL)/(IU/kg)] for FVIII:C.
Following infusion of both A-SD and A-SD/HT, an increase in the size of VWF multimers was seen and persisted for at least 24 hours. The shortening of the BT was transient, lasting less than 6 hours following treatment and did not correlate with the presence of large and intermediate size VWF multimers.26
14. CLINICAL STUDIES
Prophylaxis for Elective Surgery
Thirty seven subjects with VWD (6 Type 1, 16 Type 2A, 3 Type 2B, 12 Type 3) underwent 59 surgical procedures that included 20 dental, 7 orthopedic, 8 gastrointestinal, 6 gastrointestinal (diagnostic), 9 vascular, 3 gynecologic, 2 genitourinary, 2 dermatologic and 2 head and neck procedures administering A-SD or A-SD/HT (21 subjects were administered A-SD and 18 were administered A-SD/HT, 2 received both products) for bleeding prophylaxis (see Table 4). Prior to each surgical procedure, the investigators provided an estimation of the expected blood loss during surgery for a normal person of the same sex and of similar stature and age as the subject undergoing the same type of surgical procedure. An initial preoperative infusion of 60 VWF:RCof IU/kg (75 VWF:RCof/kg for patients less than 18 years of age), was administered one hour preoperatively. A sample was obtained 15 minutes after the initial infusion for the determination of the plasma FVIII:C level. The level had to equal or exceed 100% of normal for an operation to proceed. No cryoprecipitate or alternative FVIII product was administered during these surgical procedures. Platelets were required in only two subjects. Intra-operative infusions of A-SD and A-SD/HT at 60 VWF:RCof IU/kg (75 VWF:RCof IU/kg for patients less than 18 years of age) was administered according to the judgment of the investigator.
|
^ Two patients received both preparations; the total number of subjects is therefore less than the sum of the columns. |
|||
| Treatment | |||
| Type of Surgical Procedure | A-SD | A-SD/HT | Total |
| Number of Subjects | 21 | 18 | 37^ |
| Dental | 14 | 6 | 20 |
| Dermatologic | 1 | 1 | 2 |
| Gastrointestinal | 4 | 4 | 8 |
| Gastrointestinal (diagnostic) | 6 | 0 | 6 |
| Genitourinary | 0 | 2 | 2 |
| Gynecologic | 2 | 1 | 3 |
| Head and neck | 1 | 1 | 2 |
| Orthopedic | 4 | 3 | 7 |
| Vascular | 3 | 6 | 9 |
| Total number of procedures | 35 | 24 | 59 |
Postoperative infusions at doses of 40 to 60 VWF:RCof IU/kg (50 to 75 VWF:RCof IU/kg for pediatric patients) was administered at 8- to 12-hour intervals until healing had occurred. After achieving primary hemostasis, for maintenance of secondary hemostasis the dose was reduced after the third postoperative day. See DOSAGE AND ADMINISTRATION (2.2).
Overall, in 55 surgical procedures undertaken with a prolonged BT pre-infusion, the BT at 30 minutes post-infusion was fully corrected in 18 (32.7%) cases, partially corrected in 24 (43.6%) cases, demonstrated no correction in 12 (21.8%) cases, and was not done in one case (1.8%).
The mean blood loss was lower than predicted prospectively. Bleeding exceeding the predicted value did not correlate with correction of the BT. Three patients had bleeding which exceeded by more than 50 mL the amount predicted prospectively. Among the latter subjects, the BT 30 minutes post-infusion was normal in one and only slightly lengthened in two cases.
Surgical infusion summary data are included in Table 5.
|
* Two subjects received both products |
|||
| A-SD | A-SD/HT | Total | |
| Number of patients | 21 | 18 | 37* |
| Number of surgical procedures | 35 | 24 | 59 |
| Median number of infusions per surgical procedure (range) | 3 (1-13) | 4 (1 – 18) | 4 (1-18) |
| Median dosage VWF:RCof IU/kg | |||
| Infusion #1 (range) | 59.8 (19.8-75.1) | 59.9 (40.6 – 75.0) | 59.9 (19.8-75.1) |
| Infusion ≥ #2 combined (range) | 40.0 (4.5-75.1) | 40.0 (10.0 – 63.1) | 40.0 (4.5-75.1) |
Additionally, the surgeries were categorized as major, minor or invasive procedures according to definitions used in the study. The outcome of each surgery was evaluated according to a clinical rating scale (excellent, good, poor or none) and was considered successful if the outcome was excellent or good. These outcomes are presented in Table 6.
|
Procedure: 1=Minor, 2=Major, 3=Invasive |
||||||||||||
|
Absolute frequency & proportion of successful outcomes = 22/24 (91.66%) |
||||||||||||
|
95% Confidence Interval (CI) for the proportion of subjects with successful prophylaxis = 0.7300 to 0.9897 |
||||||||||||
| Investigator's Outcome Evaluation | Type of von Willebrand Disease | |||||||||||
| Type 1
(4 Subjects, 4 Procedures) | Type 2
(9 Subjects, 13 Procedures) | Type 3
(5 Subjects, 7 Procedures) | Total
(18 Subjects, 24 Procedures) |
|||||||||
| Procedure | Procedure | Procedure | Procedure | |||||||||
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |
| Excellent | 1 | 0 | 2 | 5 | 1 | 5 | 5 | 0 | 1 | 11 | 1 | 8 |
| Good | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 |
| Poor | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| None | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 2 | 0 |
The study results were also evaluated independently by two referees with clinical experience in this field in the same way (surgery categorization and outcome of each surgery according to a clinical rating scale).
The results for the effect of treatment on surgical prophylaxis (Referee Evaluation) per treated subject are summarized in Table 7. There is a high level of agreement between the referee evaluations and the analyzed outcome data, with a decrease of only a single success (21/24 vs. 22/24).
|
* 95% confidence interval for the proportion of subjects with successful prophylaxis, exact estimation. |
||
| Referee 1 | Referee 2 | |
| Number of Treated Subjects | 18 | 18 |
| Number of Treated Events | 24 | 24 |
| Success Absolute Frequency & Proportion (%) | 22 (0.9166) | 21 (0.8750) |
| * 95% CI for the Proportion | 0.7300 to 0.9897 | 0.6763 to 0.9734 |
A retrospective study was performed to assess the efficacy of Alphanate® (A-SD/HT) as replacement therapy in preventing excessive bleeding in subjects with congenital VWD undergoing surgical or invasive procedures, for whom DDAVP® was ineffective or inadequate. The study was performed between September 2004 and December 2005, and 61 surgeries/procedures (in 39 subjects) were evaluated.
Of the 39 subjects, 18 had Type 1 VWD (46.2%); 12 subjects (30.8%) had Type 2 VWD, and 9 subjects (23.1%) had Type 3 VWD. The median age for subjects overall was 40 years; approximately one-half of the subjects overall were male.
The primary efficacy variable was the overall treatment outcome for each surgical or invasive procedure, as rated by the investigator using a 4-point verbal rating scale (VRS): “excellent,” “good,” “poor,” or “none.” The categorization of the replacement treatment outcome according to the proposed scale was based upon the investigator's clinical experience.
The secondary efficacy variables were:
- Daily (Day 0 and Day 1) treatment outcome for each surgical or invasive procedure, rated by the investigator using the same 4-point VRS used for the primary efficacy variable. Day 0 was the day of surgery, and Day 1 was the day following surgery.
- Overall treatment outcome for each surgical or invasive procedure, rated by an independent referee committee using the same 4-point VRS used for the primary efficacy variable.
In addition, an independent referee committee was convened to evaluate the efficacy outcomes. The committee was composed of 2 physicians with demonstrated clinical expertise treating subjects with similar medical characteristics to those of the study population. The committee was blinded to the investigator ratings; and each referee evaluated the outcomes independent of one another.
More than 90% received an investigator and referee's overall and daily rating of “effective” (“excellent” or “good”). The results of the primary efficacy analysis are in Table 8.
|
a Binomial test (H0: < 70% of procedures have an overall rating of effective). |
|||
|
b Effective = Investigator rating of “excellent” or “good.” |
|||
|
c Non-effective = Investigator rating of “poor” or “none.” |
|||
| Outcome of Alphanate Treatment | Proportion of Procedures (%) | 95% Confidence Interval | P Valuea |
| Effectiveb | 95.1 | 87.8 – 98.6 | < 0.0001 |
| Non-effectivec | 4.9 | 1.4 – 12.2 | |
The results of the analysis of daily investigator ratings are in Table 9.
|
a Study Day 0 = day of surgery. |
||||
|
b Binomial test (H0: < 70% of procedures have an overall rating of effective). |
||||
|
c Effective = Investigator rating of “excellent” or “good.” |
||||
|
d Non-effective = Investigator rating of “poor” or “none.” |
||||
| Study Daya | Outcome of Alphanate® Treatment | Proportion of Procedures (%) | 95% Confidence Interval | P Valueb |
| 0 | Effectivec | 95.1 | 87.8 – 98.6 | < 0.0001 |
| Non-effectived | 4.9 | 1.4 – 12.2 | ||
| 1 | Effective | 91.8 | 83.5 – 96.7 | < 0.0001 |
| Non-effective | 8.2 | 3.3 – 16.5 | ||
The results of the analysis of overall referee ratings are in Table 10.
|
a Binomial test (H0: < 70% of procedures have an overall rating of effective). |
|||
|
b Effective = Referee rating of “excellent” or “good.” |
|||
|
c Non-effective = Referee rating of “poor” or “none.” |
|||
| Outcome of Alphanate® Treatment | Proportion of Procedures (%) | 95% Confidence Interval | P Valuea |
| Effectiveb | 91.8 | 83.5 – 96.7 | < 0.0001 |
| Non-effectivec | 8.2 | 3.3 – 16.5 | |
The overall investigator ratings are summarized by type of VWD in Table 11.
|
a Minor surgery also includes invasive procedures. |
||||||||
| Investigator's Overall Rating | Type of von Willebrand Disease | |||||||
| Type 1
(18 Subjects, 22 Procedures) | Type 2
(12 Subjects, 23 Procedures) | Type 3
(9 Subjects, 16 Procedures) | Total
(39 Subjects, 61 Procedures) |
|||||
| Major | Minora | Major | Minor | Major | Minor | Major | Minor | |
| Excellent | 6 (85.7%) | 12 (80.0%) | 2 (50.0%) | 18 (94.7%) | 0 (0.0%) | 13 (86.7%) | 8 (66.7%) | 43 (87.8%) |
| Good | 1 (14.3%) | 3 (20.0%) | 2 (50.0%) | 0 (0.0%) | 0 (0.0%) | 1 (6.7%) | 3 (25.0%) | 4 (8.2%) |
| Poor | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (5.3%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (2.0%) |
| None | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (100) | 1 (6.7%) | 1 (8.3%) | 1 (2.0%) |
The majority of ratings were “excellent” (≥ 81.3% in each VWD type). Only 2 procedures in 1 subject with Type 3 VWD received an overall efficacy rating of “none,” and 1 procedure in 1 subject with Type 2 VWD received an overall efficacy rating of “poor.”
The total dose of Alphanate® received over the entire perioperative period of the retrospective study is summarized in Table 12.
| A-SD/HT | |
| Number of patients | 39 |
| Number of surgical procedures | 61 |
| Mean number of infusions | 5.9 |
| Median number of infusions per surgical procedure (range) | 3 (1-27) |
15. REFERENCES
- Eyster, M.E. Hemophilia: A Guide for the Primary Care Physician. Postgrad Med 1978; 64:75-81.
- Shanbrom, E., Thelin, M. Experimental Prophylaxis of Severe Hemophilia with a Factor VIII Concentrate. JAMA 1969; 208(9):1853-1856.
- Levine, P.H. Hemophilia and Allied Conditions. In: Brain, M.C. ed. Current Therapy in Hematology-Oncology: 1983-1984, New York: BC Decker, 1983, pp. 147-152.
- Rizza, C.R., Biggs, R. Blood Products in the Management of Haemophilia and Christmas Disease. In: Poller, L., ed. Recent Advances in Blood Coagulation, Boston: Little Brown, 1969, pp. 179-195.
- Hathaway, W.E., Mahasandana, C., Clarke, S. Alteration of Platelet Function After Transfusion in Hemophilia. Proc 14th Ann Mtg, Am Soc Hematol 1971, Abstracts, 58, No. 88.
- Kasper, C.K. Incidence and Course of Inhibitors Among Patients with classic Hemophilia. Thromb Diath Haemorrh 1973; 30:263-271.
- Rizza, C.R., Biggs, R. The Treatment of Patients Who Have Factor VIII Antibodies Br J Haematol 1973; 24:65-82.
- Roberts, H.R., Knowles, M.R., Jones, T.L., McMillan, C. The Use of Factor VIII in the Management of Patients with Factor VIII Inhibitors. In: Brinkhous, K.M., ed. Hemophilia and New Hemorrhagic States, International Symposium, New York, University of North Carolina Press, 1970, pp. 152-163.
- Federici, A.B., Baudo, F., Caracciolo, C., Mancuso, G., Mazzucconi, M.G., Musso, R., Schinco, P.C., Targhetta. R., Mannucci, P.M.. Clinical efficacy of highly purified, doubly virus-inactivated factor VIII/von Willebrand factor concentrate (Fanhdi®) in the treatment of von Willebrand disease: a retrospective clinical study. Haemophilia 2002; 8:761-767.
- Federici, A.B. Managment of von Willebrand disease with FVIII/von Willebrand factor concentrates: results from current studies and surveys. Blood Coagul Fibrinolysis 2005;16(Suppl 1):S17-S21.
- Mannucci, P.M. How I treat patients with von Willebrand disease. Blood 2001; 97:1915-1919.
- Mannucci, P.M. Treatment of von Willebrand's Disease. N Engl J Med 2004;351:683-694.
- Mannucci, P.M. Venous Thromboembolism in von Willebrand Disease. Thromb Haemost 2002; 88:378-379.
- Markis, M., Colvin, B., Gupta, V., Shields, M.L., Smith, M.P. Venous Thrombosis Following the Use of Intermediate Purity FVIII Concentrate to Treat Patients with von Willebrand Disease. Thromb Haemost 2002; 88: 387-388.
- Biggs, R. Jaundice and Antibodies Directed Against Factors VIII and IX in Patients Treated for Haemophilia or Christmas Disease in the United Kingdom. Br J Haematol 1974; 26:313-329.
- Kasper, C.K., Kipnis, S.A. Hepatitis and Clotting Factor Concentrates. JAMA1972; 221:510.
- Mannucci, P.M., Federici, A.B. Antibodies to von Willebrand Factor in von Willebrand Disease. In: Aledort L.M., Hoyer L.W., Reisener J.M., White II, G.C. eds. Inhibitors to coagulation factor in the 1990s, 1995, Plenum Press, pp. 87-92.
- Data on file at Grifols Biologicals Inc.
- Fujimura, Y., Titani, K., Holland, L.Z., Roberts, J.R., Kostel, P., Ruggeri, Z.M., Zimmerman, T.S. A heparin-binding domain of human von Willebrand factor: Characterization and localization to a tryptic fragment extending from amino acid residue Val-449 to Lys-728. J Biol Chem 1987; 262(4):1734-1739.
- Horowitz, B. Investigations into the Application of Tri (n-Butyl) Phosphate/Detergent Mixture to Blood Derivatives. In: Morgenthaler, J-J ed. Viral Inactivation in Plasma Products, Karger, 1989, 56:83-96.
- Edwards, C.A., Piet, M.P.J., Chin, S., Horowitz, B. Tri (n-Butyl) Phosphate/Detergent Treatment of Licensed Therapeutic and Experimental Blood Derivatives. Vox Sang 1987; 52:53-59.
- Hershgold, E.J. Properties of Factor VIII (Antihaemophilic Factor). In: Spaet, T.H., ed. Progress in Hemostasis and Thrombosis, Grune and Stratton Publisher, 1974, 2:99-139.
- Ashenhurst, J.B., Langehenning, P.L., Seeler, R.A. Early Treatment of Bleeding Episodes with l0 U/Kg of Factor VIII. Blood 1977:50:181.
- Hoyer, L.W. The Factor VIII complex: Structure and function. Blood 1981; 58:1-13.
- Meyer, D., and Girma, J-P. von Willebrand factor: Structure and function. Thromb Haemost 1983; 70:99-104.
- Mannucci, P.M., Chediak, J., Hanna, W. Byrnes, J.J., Kessler, C.M, Ledford, M., Retzios, A.D., Kapelan, B.A., Gallagher, P., Schwartz, R.S., and the Alphanate Study Group. Treatment of von Willebrand's Disease (VWD) with a high purity factor VIII concentrate: Dissociation between correction of the bleeding time (BT), VWF multimer pattern, and treatment efficacy. Blood 1999; 94 (Suppl 1, Part 2 of 2):98b.
16. HOW SUPPLIED/STORAGE AND HANDLING
Alphanate® is supplied in sterile, lyophilized form in a single dose vial with a vial of diluent (Sterile Water for Injection, USP), a double-ended transfer needle and microaggregate filter for use in administration. International unit activity of Factor VIII and VWF:RCof are stated on the carton and label of each vial. It is available in the following potencies:
- 250 IU/5 mL single dose vial (NDC 68516-4601-1)
- 500 IU/5 mL single dose vial (NDC 68516-4602-1)
- 1000 IU/10 mL single dose vial (NDC 68516-4603-2)
- 1500 IU/10 mL single dose vial (NDC 68516-4604-2)
Storage
Alphanate® should be stored at temperatures between 2 and 8 °C. Do not freeze to prevent damage to diluent vial. Alphanate® may be stored at room temperature not to exceed 30 ºC for up to 2 months. When removed from refrigeration, record the date removed on the space provided on the carton.
17. PATIENT COUNSELING INFORMATION
Patients should be informed of the early symptoms and signs of hypersensitivity reaction, including hives, generalized urticaria, chest tightness, dyspnea, wheezing, faintness, hypotension, and anaphylaxis. Patients should be advised to discontinue use of the product and contact their physician and/or seek immediate emergency care, depending on the severity of the reaction, if these symptoms occur. It is recommended that the lot number of the vials used be recorded when Alphanate® is administered.
17.1 Thromboembolic Events
Thromboembolic events have been reported in von Willebrand Disease patients receiving Antihemophilic Factor/von Willebrand Factor Complex replacement therapy, especially in the setting of known risk factors for thrombosis.13,14 Early reports might indicate a higher incidence in females. In addition, endogenous high levels of FVIII have also been associated with thrombosis but no causal relationship has been established. In all VWD patients in situations of high thrombotic risk receiving coagulation factor replacement therapy, caution should be exercised and antithrombotic measures should be considered. See also WARNINGS AND PRECAUTIONS (5.1).
17.2 Infections
Because Antihemophilic Factor/von Willebrand Factor Complex (Human), Alphanate® is made from pooled human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, and theoretically, the Creutzfeldt-Jakob disease (CJD) agent. Stringent procedures designed to reduce the risk of adventitious agent transmission have been employed in the manufacture of this product, from the screening of plasma donors and the collection and testing of plasma, through the application of viral elimination/reduction steps such as solvent detergent and heat treatment in the manufacturing process. Despite these measures, such products can still potentially transmit disease; therefore, the risk of infectious agents cannot be totally eliminated. All infections thought by a physician possibly to have been transmitted by this product should be reported to the manufacturer at 888-GRIFOLS (888-474-3657) for USA and 323-225-2221 for international. The physician should weigh the risks and benefits of the use of this product and should discuss these with the patient. See also WARNINGS AND PRECAUTIONS (5.2).
17.3 Inhibitor Formation
Some patients develop inhibitors to Factor VIII. These inhibitors are circulating antibodies (i.e., globulins) that neutralize the procoagulant activity of Factor VIII. No studies have been conducted with Alphanate® to evaluate inhibitor formation. Therefore, it is not known whether there are greater, lesser or the same risks of developing inhibitors due to the use of this product than there are with other antihemophilic factor preparations. Patients with these inhibitors may not respond to treatment with Antihemophilic Factor/von Willebrand Factor Complex (Human), or the response may be much less than would otherwise be expected; therefore, larger doses of Antihemophilic Factor/von Willebrand Factor Complex (Human) are often required. The management of bleeding in patients with inhibitors requires careful monitoring, especially if surgical procedures are indicated.6-8 See also WARNINGS AND PRECAUTIONS (5.3).
Manufactured and Distributed by:
Grifols Biologicals Inc.
Los Angeles, CA 90032, U.S.A.
U. S. License No. 1694
DATE OF REVISION: 01/2007
08-8181
Principal Display Panel Information – Alphanate®
PACKAGE LABEL – PRINCIPAL DISPLAY PANEL – 250 IU VIAL
NDC 68516-4601-1
Antihemophilic Factor/von Willebrand Factor Complex (Human) Alphanate®
Solvent Detergent/Heat Treated 5 mL
Storage : Store between 2 and 8 °C. May be stored at room temperature not to exceed 30 °C for up to 2 months.
Rx only. Single dose container for intravenous administration only.
Grifols Biologicals Inc.
Los Angeles, CA 90032, USA
U.S. License No. 1694
GRIFOLS

PACKAGE LABEL – PRINCIPAL DISPLAY PANEL – 500 IU VIAL
NDC 68516-4602-1
Antihemophilic Factor/von Willebrand Factor Complex (Human) Alphanate®
Solvent Detergent/Heat Treated 5 mL
Storage : Store between 2 and 8 °C. May be stored at room temperature not to exceed 30 °C for up to 2 months.
Rx only. Single dose container for intravenous administration only.
Grifols Biologicals Inc.
Los Angeles, CA 90032, USA
U.S. License No. 1694
GRIFOLS

PACKAGE LABEL – PRINCIPAL DISPLAY PANEL – 1000 IU VIAL
NDC 68516-4603-2
Antihemophilic Factor/von Willebrand Factor Complex (Human) Alphanate®
Solvent Detergent/Heat Treated 10 mL
Storage : Store between 2 and 8 °C. May be stored at room temperature not to exceed 30 °C for up to 2 months.
Rx only. Single dose container for intravenous administration only.
Grifols Biologicals Inc.
Los Angeles, CA 90032, USA
U.S. License No. 1694
GRIFOLS

PACKAGE LABEL – PRINCIPAL DISPLAY PANEL – 1500 IU VIAL
NDC 68516-4604-2
Antihemophilic Factor/von Willebrand Factor Complex (Human) Alphanate®
Solvent Detergent/Heat Treated 10 mL
Storage : Store between 2 and 8 °C. May be stored at room temperature not to exceed 30 °C for up to 2 months.
Rx only. Single dose container for intravenous administration only.
Grifols Biologicals Inc.
Los Angeles, CA 90032, USA
U.S. License No. 1694
GRIFOLS

PACKAGE LABEL – PRINCIPAL DISPLAY PANEL – 250 IU CARTON
GRIFOLS
NDC 68516-4601-1
Antihemophilic Factor/von Willebrand Factor Complex (Human)
Alphanate®
Solvent Detergent / Heat Treated
5 mL

PACKAGE LABEL – PRINCIPAL DISPLAY PANEL – 500 IU CARTON
GRIFOLS
NDC 68516-4602-1
Antihemophilic Factor/von Willebrand Factor Complex (Human)
Alphanate®
Solvent Detergent / Heat Treated
5 mL

PACKAGE LABEL – PRINCIPAL DISPLAY PANEL – 1000 IU CARTON
GRIFOLS
NDC 68516-4603-2
Antihemophilic Factor/von Willebrand Factor Complex (Human)
Alphanate®
Solvent Detergent / Heat Treated
10 mL

PACKAGE LABEL – PRINCIPAL DISPLAY PANEL – 1500 IU CARTON
GRIFOLS
NDC 68516-4604-2
Antihemophilic Factor/von Willebrand Factor Complex (Human)
Alphanate®
Solvent Detergent / Heat Treated
10 mL

| ALPHANATE
antihemophilic factor/von willebrand factor complex (human) kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA102475 | 01/31/2007 | |
| ALPHANATE
antihemophilic factor/von willebrand factor complex (human) kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA102475 | 01/31/2007 | |
| ALPHANATE
antihemophilic factor/von willebrand factor complex (human) kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA102475 | 01/31/2007 | |
| ALPHANATE
antihemophilic factor/von willebrand factor complex (human) kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA102475 | 01/31/2007 | |
| Labeler - Grifols Biologicals Inc. (096019096) |
| Registrant - Grifols Biologicals Inc. (096019096) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Grifols Biologicals Inc. | 092694538 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Grifols Biologicals Inc. | 121076871 | MANUFACTURE | |