REVLIMID
-
lenalidomide capsule
Celgene Corporation
----------
REVLIMID® (lenalidomide)
5 mg, 10 mg, 15 mg and 25 mg capsules
WARNINGS:
- POTENTIAL FOR HUMAN BIRTH DEFECTS
- HEMATOLOGIC TOXICITY (NEUTROPENIA AND THROMBOCYTOPENIA)
- DEEP VENOUS THROMBOSIS AND PULMONARY EMBOLISM
POTENTIAL FOR HUMAN BIRTH DEFECTS
WARNING: POTENTIAL FOR HUMAN BIRTH DEFECTS
LENALIDOMIDE IS AN ANALOGUE OF THALIDOMIDE. THALIDOMIDE IS A KNOWN HUMAN TERATOGEN THAT CAUSES SEVERE LIFE-THREATENING HUMAN BIRTH DEFECTS. IF LENALIDOMIDE IS TAKEN DURING PREGNANCY, IT MAY CAUSE BIRTH DEFECTS OR DEATH TO AN UNBORN BABY. FEMALES SHOULD BE ADVISED TO AVOID PREGNANCY WHILE TAKING REVLIMID® (lenalidomide).
Special Prescribing Requirements
BECAUSE OF THIS POTENTIAL TOXICITY AND TO AVOID FETAL EXPOSURE TO REVLIMID® (lenalidomide), REVLIMID® (lenalidomide) IS ONLY AVAILABLE UNDER A SPECIAL RESTRICTED DISTRIBUTION PROGRAM. THIS PROGRAM IS CALLED "RevAssist®". UNDER THIS PROGRAM, ONLY PRESCRIBERS AND PHARMACISTS REGISTERED WITH THE PROGRAM CAN PRESCRIBE AND DISPENSE THE PRODUCT. IN ADDITION, REVLIMID® (lenalidomide) MUST ONLY BE DISPENSED TO PATIENTS WHO ARE REGISTERED AND MEET ALL THE CONDITIONS OF THE RevAssist® PROGRAM.
PLEASE SEE THE FOLLOWING INFORMATION FOR PRESCRIBERS, FEMALE PATIENTS, AND MALE PATIENTS ABOUT THIS RESTRICTED DISTRIBUTION PROGRAM.
RevAssist® PROGRAM DESCRIPTION
Prescribers
REVLIMID® (lenalidomide) can be prescribed only by licensed prescribers who are registered in the RevAssist® program and understand the potential risk of teratogenicity if lenalidomide is used during pregnancy.
Effective contraception must be used by female patients of childbearing potential for at least 4 weeks before beginning REVLIMID® (lenalidomide) therapy, during REVLIMID® (lenalidomide) therapy, during dose interruptions and for 4 weeks following discontinuation of REVLIMID® (lenalidomide) therapy. Reliable contraception is indicated even where there has been a history of infertility, unless due to hysterectomy or because the patient has been postmenopausal naturally for at least 24 consecutive months. Two reliable forms of contraception must be used simultaneously unless continuous abstinence from heterosexual sexual contact is the chosen method. Females of childbearing potential should be referred to a qualified provider of contraceptive methods, if needed. Sexually mature females who have not undergone a hysterectomy, have not had a bilateral oophorectomy or who have not been postmenopausal naturally for at least 24 consecutive months (i.e., who have had menses at some time in the preceding 24 consecutive months) are considered to be females of childbearing potential.
Before prescribing REVLIMID® (lenalidomide), females of childbearing potential should have 2 negative pregnancy tests (sensitivity of at least 50 mIU/mL). The first test should be performed within 10–14 days, and the second test within 24 hours prior to prescribing REVLIMID® (lenalidomide). A prescription for REVLIMID® (lenalidomide) for a female of childbearing potential must not be issued by the prescriber until negative pregnancy tests have been verified by the prescriber.
Male Patients: It is not known whether lenalidomide is present in the semen of patients receiving the drug. Therefore, males receiving REVLIMID® (lenalidomide) must always use a latex condom during any sexual contact with females of childbearing potential even if they have undergone a successful vasectomy.
Once treatment has started and during dose interruptions, pregnancy testing for females of childbearing potential should occur weekly during the first 4 weeks of use, then pregnancy testing should be repeated every 4 weeks in females with regular menstrual cycles. If menstrual cycles are irregular, the pregnancy testing should occur every 2 weeks. Pregnancy testing and counseling should be performed if a patient misses her period or if there is any abnormality in her pregnancy test or in her menstrual bleeding. REVLIMID® (lenalidomide) treatment must be discontinued during this evaluation.
Pregnancy test results should be verified by the prescriber and the pharmacist prior to dispensing any prescription.
If pregnancy does occur during REVLIMID® (lenalidomide) treatment, REVLIMID® (lenalidomide) must be discontinued immediately.
Any suspected fetal exposure to REVLIMID® (lenalidomide) should be reported to the FDA via the MedWatch number at 1-800-FDA-1088 and also to Celgene Corporation at 1-888-423-5436. The patient should be referred to an obstetrician/gynecologist experienced in reproductive toxicity for further evaluation and counseling.
Female Patients
REVLIMID® (lenalidomide) should be used in females of childbearing potential only when the patient MEETS ALL OF THE FOLLOWING CONDITIONS (i.e., she is unable to become pregnant while on lenalidomide therapy):
- she understands and can reliably carry out instructions.
- she is capable of complying with the mandatory contraceptive measures, pregnancy testing, patient registration, and patient survey as described in the RevAssist® program.
- she has received and understands both oral and written warnings of the potential risks of taking lenalidomide during pregnancy and of exposing a fetus to the drug.
- she has received both oral and written warnings of the risk of possible contraception failure and of the need to use two reliable forms of contraception simultaneously, unless continuous abstinence from heterosexual sexual contact is the chosen method. Sexually mature females who have not undergone a hysterectomy or who have not been postmenopausal for at least 24 consecutive months (i.e., who have had menses at some time in the preceding 24 consecutive months), or had a bilateral oophorectomy are considered to be females of childbearing potential.
- she acknowledges, in writing, her understanding of these warnings and of the need for using two reliable methods of contraception for 4 weeks prior to beginning lenalidomide therapy, during lenalidomide therapy, during dose interruptions and for 4 weeks after discontinuation of lenalidomide therapy.
- she has had two negative pregnancy tests with a sensitivity of at least 50 mIU/mL, within 10-14 days and 24 hours prior to beginning therapy.
- if the patient is between 12 and 18 years of age, her parent or legal guardian must have read the educational materials and agreed to ensure compliance with the above.
Male Patients
REVLIMID® (lenalidomide) should be used in sexually active males when the PATIENT MEETS ALL OF THE FOLLOWING CONDITIONS:
- he understands and can reliably carry out instructions.
- he is capable of complying with the mandatory contraceptive measures that are appropriate for men, patient registration, and patient survey as described in the RevAssist® program.
- he has received and understands both oral and written warnings of the potential risks of taking lenalidomide and exposing a fetus to the drug.
- he has received both oral and written warnings of the risk of possible contraception failure and that it is unknown whether lenalidomide is present in semen. He has been instructed that he must always use a latex condom during any sexual contact with females of childbearing potential, even if he has undergone a successful vasectomy.
- he acknowledges, in writing, his understanding of these warnings and of the need to use a latex condom during any sexual contact with females of childbearing potential, even if he has undergone a successful vasectomy. Females of childbearing potential are considered to be sexually mature females who have not undergone a hysterectomy, have not had a bilateral oophorectomy or who have not been postmenopausal for at least 24 consecutive months (i.e., who have had menses at any time in the preceding 24 consecutive months).
- if the patient is between 12 and 18 years of age, his parent or legal guardian must have read the educational materials and agreed to ensure compliance with the above.
HEMATOLOGIC TOXICITY (NEUTROPENIA AND THROMBOCYTOPENIA)
This drug is associated with significant neutropenia and thrombocytopenia. Eighty percent of patients with del 5q myelodysplastic syndromes had to have a dose delay/reduction during the major study. Thirty-four percent of patients had to have a second dose delay/reduction. Grade 3 or 4 hematologic toxicity was seen in 80% of patients enrolled in the study. Patients on therapy for del 5q myelodysplastic syndromes should have their complete blood counts monitored weekly for the first 8 weeks of therapy and at least monthly thereafter. Patients may require dose interruption and/or reduction. Patients may require use of blood product support and/or growth factors. (See DOSAGE AND ADMINISTRATION)
DEEP VENOUS THROMBOSIS AND PULMONARY EMBOLISM
This drug has demonstrated a significantly increased risk of deep venous thrombosis (DVT) and pulmonary embolism (PE) in patients with multiple myeloma who were treated with REVLIMID® (lenalidomide) combination therapy. Patients and physicians are advised to be observant for the signs and symptoms of thromboembolism. Patients should be instructed to seek medical care if they develop symptoms such as shortness of breath, chest pain, or arm or leg swelling. It is not known whether prophylactic anticoagulation or antiplatelet therapy prescribed in conjunction with REVLIMID® (lenalidomide) may lessen the potential for venous thromboembolic events. The decision to take prophylactic measures should be done carefully after an assessment of an individual patient’s underlying risk factors.
You can get the information about REVLIMID® (lenalidomide) and the RevAssist® program on the internet at www.REVLIMID.com or by calling the manufacturer’s toll free number 1-888-423-5436.
DESCRIPTION
REVLIMID® (lenalidomide), a thalidomide analogue, is an immunomodulatory agent with antiangiogenic and antineoplastic properties. The chemical name is 3-(4-amino-1-oxo 1,3-dihydro-2H-isoindol-2-yl) piperidine-2,6-dione and it has the following chemical structure:
3-(4-amino-1-oxo 1,3-dihydro-2H-isoindol-2-yl) piperidine-2,6-dione
The empirical formula for lenalidomide is C13H13N3O3, and the gram molecular weight is 259.3.
Lenalidomide is an off-white to pale-yellow solid powder. It is soluble in organic solvent/water mixtures, and buffered aqueous solvents. Lenalidomide is more soluble in organic solvents and low pH solutions. Solubility was significantly lower in less acidic buffers, ranging from about 0.4 to 0.5 mg/ml. Lenalidomide has an asymmetric carbon atom and can exist as the optically active forms S(-) and R(+), and is produced as a racemic mixture with a net optical rotation of zero.
REVLIMID® (lenalidomide) is available in 5 mg, 10 mg, 15 mg and 25 mg capsules for oral administration. Each capsule contains lenalidomide as the active ingredient and the following inactive ingredients: lactose anhydrous, microcrystalline cellulose, croscarmellose sodium, and magnesium stearate. The 5 mg and 25 mg capsule shell contains gelatin, titanium dioxide and black ink. The 10 mg capsule shell contains gelatin, FD&C blue #2, yellow iron oxide, titanium dioxide and black ink. The 15 mg capsule shell contains gelatin, FD&C blue #2, titanium dioxide and black ink.
CLINICAL PHARMACOLOGY
Mechanism of Action
The mechanism of action of lenalidomide remains to be fully characterized. Lenalidomide possesses antineoplastic, immunomodulatory and antiangiogenic properties. Lenalidomide inhibited the secretion of pro-inflammatory cytokines and increased the secretion of antiinflammatory cytokines from peripheral blood mononuclear cells. Lenalidomide inhibited cell proliferation with varying effectiveness (IC50s) in some but not all cell lines. Of cell lines tested, lenalidomide was effective in inhibiting growth of Namalwa cells (a human B cell lymphoma cell line with a deletion of one chromosome 5) but was much less effective in inhibiting growth of KG-1 cells (human myeloblastic cell line, also with a deletion of one chromosome 5) and other cell lines without chromosome 5 deletions. Lenalidomide inhibited the growth of multiple myeloma cells from patients, as well as MM.1S cells (a human multiple myeloma cell line), by inducing cell cycle arrest and apoptosis.
Lenalidomide inhibited the expression of cyclooxygenase-2 (COX-2) but not COX-1 in vitro.
Pharmacokinetics and Drug Metabolism
Absorption:
Lenalidomide, in healthy volunteers, is rapidly absorbed following oral administration with maximum plasma concentrations occurring between 0.625 and 1.5 hours post-dose. Co‑administration with food does not alter the extent of absorption (AUC) but does reduce the maximal plasma concentration (Cmax) by 36%. The pharmacokinetic disposition of lenalidomide is linear. Cmax and AUC increase proportionately with increases in dose. Multiple dosing at the recommended dose-regimen does not result in drug accumulation.
Pharmacokinetic sampling in myelodysplastic syndromes (MDS) patients was not performed. In multiple myeloma patients maximum plasma concentrations occurred between 0.5 and 4.0 hours post-dose both on Days 1 and 28. AUC and Cmax values increase proportionally with dose following single and multiple doses. Exposure (AUC) in multiple myeloma patients is 57% higher than in healthy male volunteers.
Pharmacokinetic Parameters
Distribution:
In vitro (14C)-lenalidomide binding to plasma proteins is approximately 30%.
Metabolism and Excretion:
The metabolic profile of lenalidomide in humans has not been studied. In healthy volunteers, approximately two-thirds of lenalidomide is eliminated unchanged through urinary excretion. The process exceeds the glomerular filtration rate and therefore is partially or entirely active. Half‑life of elimination is approximately 3 hours.
Special Populations:
Patients with Renal Insufficiency: The pharmacokinetics of lenalidomide were studied in patients with renal impairment due to nonmalignant conditions. In this study, 5 patients with mild renal function impairment (creatinine clearance 57-74 mL/min), 6 patients with moderate renal function impairment (creatinine clearance 33-46 mL/min), 6 patients with severe renal function impairment (creatinine clearance 17-29 mL/min), and 6 patients with end stage renal disease requiring dialysis were administered a single oral 25-mg dose of REVLIMID® (lenalidomide). As a control group comparator, 7 healthy subjects of similar age with normal renal function (creatinine clearance 83-145 mL/min) were also administered a single oral 25-mg dose of REVLIMID® (lenalidomide). As creatinine clearance decreased from mild to severe impairment, half-life increased and drug clearance decreased linearly. Patients with moderate and severe renal impairment had a 3-fold increase in half-life and a 66% to 75% decrease in drug clearance compared to healthy subjects. Patients on hemodialysis (n=6) given a single, 25-mg dose of lenalidomide had an approximate 4.5-fold increase in half-life and an 80% decrease in drug clearance compared to healthy subjects. Approximately 40% of the administered dose was removed from the body during a single dialysis session.
Adjustment of the starting dose of REVLIMID® (lenalidomide) is recommended in patients with moderate or severe (CLcr < 60 mL/min) renal impairment and in patients on dialysis. See DOSAGE AND ADMINISTRATION.
In multiple myeloma patients, those patients with mild renal impairment had an AUC 56% greater than those with normal renal function.
Patients with Hepatic Disease: The pharmacokinetics of lenalidomide in patients with hepatic impairment have not been studied.
Age: The effects of age on the pharmacokinetics of lenalidomide have not been studied.
Pediatric: No pharmacokinetic data are available in patients below the age of 18 years.
Gender: The effects of gender on the pharmacokinetics of lenalidomide have not been studied.
Race: Pharmacokinetic differences due to race have not been studied.
CLINICAL STUDIES
Myelodysplastic Syndromes (MDS) with a Deletion 5q Cytogenetic Abnormality
The efficacy and safety of REVLIMID® (lenalidomide) were evaluated in patients with transfusion dependent anemia in Low- or Intermediate-1- risk MDS with a 5q (q31-33) cytogenetic abnormality in isolation or with additional cytogenetic abnormalities, at a dose of 10 mg once daily or 10 mg once daily for 21 days every 28 days in an open-label, single-arm, multi-center study. The major study was not designed nor powered to prospectively compare the efficacy of the 2 dosing regimens. Sequential dose reductions to 5 mg daily and 5 mg every other day, as well as dose delays, were allowed for toxicity.
This major study enrolled 148 patients who had RBC transfusion dependent anemia. RBC-transfusion dependence was defined as having received ≥ 2 units of RBCs within 8 weeks prior to study treatment. The study enrolled patients with absolute neutrophil counts (ANC) ≥ 500/mm3, platelet counts ≥ 50,000/mm3, serum creatinine ≤ 2.5 mg/dL, serum SGOT/AST or SGPT/ALT ≤ 3.0 x upper limit of normal (ULN), and serum direct bilirubin ≤ 2.0 mg/dL. Granulocyte colony-stimulating factor was permitted for patients who developed neutropenia or fever in association with neutropenia. Baseline patient and disease-related characteristics are summarized in Table 1.
| [a] IPSS Risk Category: Low (combined score = 0), Intermediate-1 (combined score = 0.5 to 1.0), Intermediate-2 (combined score = 1.5 to 2.0), High (combined score ≥ 2.5); Combined score = (Marrow blast score + Karyotype score + Cytopenia score) | ||
| [b] French-American-British (FAB) classification of MDS. | ||
| Overall (N=148) |
||
|---|---|---|
| Age (years) | ||
|
Median | 71.0 | |
| Min, Max | 37.0, 95.0 | |
| Gender | n | (%) |
| Male | 51 | (34.5) |
| Female | 97 | (65.5) |
| Race | n | (%) |
| White | 143 | (96.6) |
| Other | 5 | (3.4) |
| Duration of MDS (years) | ||
| Median | 2.5 | |
| Min, Max | 0.1, 20.7 | |
| Del 5 (q31-33) Cytogenetic Abnormality | n | (%) |
| Yes | 148 | (100.0) |
| Other cytogenetic abnormalities | 37 | (25.2) |
| IPSS Score [a] | n | (%) |
| Low (0) | 55 | (37.2) |
| Intermediate-1 (0.5-1.0) | 65 | (43.9) |
| Intermediate-2 (1.5-2.0) | 6 | (4.1) |
| High (≥2.5) | 2 | (1.4) |
| Missing | 20 | (13.5) |
| FAB Classification [b] from central review | n | (%) |
| RA | 77 | (52.0) |
| RARS | 16 | (10.8) |
| RAEB | 30 | (20.3) |
| CMML | 3 | (2.0) |
The frequency of RBC-transfusion independence was assessed using criteria modified from the International Working Group (IWG) response criteria for MDS. RBC transfusion independence was defined as the absence of any RBC transfusion during any consecutive “rolling” 56 days (8 weeks) during the treatment period.
Transfusion independence was seen in 99/148 (67%) patients (95% CI [59, 74]). The median duration from the date when RBC transfusion independence was first declared (i.e., the last day of the 56-day RBC transfusion-free period) to the date when an additional transfusion was received after the 56-day transfusion-free period among the 99 responders was 44 weeks (range of 0 to >67 weeks).
Ninety percent of patients who achieved a transfusion benefit did so by completion of three months in the study.
RBC-transfusion independence rates were unaffected by age or gender.
The dose of REVLIMID® (lenalidomide) was reduced or interrupted at least once due to an adverse event in 118 (79.7%) of the 148 patients; the median time to the first dose reduction or interruption was 21 days (mean, 35.1 days; range, 2-253 days), and the median duration of the first dose interruption was 22 days (mean, 28.5 days; range, 2-265 days). A second dose reduction or interruption due to adverse events was required in 50 (33.8%) of the 148 patients. The median interval between the first and second dose reduction or interruption was 51 days (mean, 59.7 days; range, 15-205 days) and the median duration of the second dose interruption was 21 days (mean, 26 days; range, 2-148 days).
Granulocyte colony-stimulating factors were permitted for patients who developed neutropenia or fever in association with neutropenia.
Multiple Myeloma
Two randomized studies (Studies 1 and 2) were conducted to evaluate the efficacy and safety of REVLIMID® (lenalidomide). These multicenter, multinational, double-blind, placebo-controlled studies compared REVLIMID® (lenalidomide) plus oral pulse high-dose dexamethasone therapy to dexamethasone therapy alone, in patients with multiple myeloma who had received at least one prior treatment.
In both studies, patients in the REVLIMID® (lenalidomide)/dexamethasone group took 25 mg of REVLIMID® (lenalidomide) orally once daily on Days 1 to 21 and a matching placebo capsule once daily on Days 22 to 28 of each 28‑day cycle. Patients in the placebo/dexamethasone group took 1 placebo capsule on Days 1 to 28 of each 28‑day cycle. Patients in both treatment groups took 40 mg of dexamethasone orally once daily on Days 1 to 4, 9 to 12, and 17 to 20 of each 28‑day cycle for the first 4 cycles of therapy.
The dose of dexamethasone was reduced to 40 mg orally once daily on Days 1 to 4 of each 28‑day cycle after the first 4 cycles of therapy. In both studies, treatment was to continue until disease progression.
In both studies, dose adjustments were allowed based on clinical and laboratory findings. Sequential dose reductions to 15 mg daily, 10 mg daily and 5 mg daily were allowed for toxicity. (See DOSAGE AND ADMINISTRATION)
Table 2 summarizes the baseline patient and disease characteristics in the two studies. In both studies, baseline demographic and disease-related characteristics were comparable between the REVLIMID® (lenalidomide)/dexamethasone and placebo/dexamethasone groups.
| Study 1 | Study 2 | |||
| REVLIMID/Dex
N=170 | Placebo/Dex
N=171 | REVLIMID/Dex
N=176 | Placebo/Dex
N=175 |
|
| Patient Characteristics | ||||
| Age (years) | ||||
| Median | 64 | 62 | 63 | 64 |
| Min,Max | 36,86 | 37, 85 | 33, 84 | 40, 82 |
| Sex | ||||
| Male | 102 (60%) | 101 (59%) | 104 (59%) | 103 (59%) |
| Female | 68 (40%) | 70 (41%) | 72 (41%) | 72 (41%) |
| Race/Ethnicity | ||||
| White | 134 (79%) | 143 (84%) | 172 (98%) | 175 (100%) |
| Other | 36 (21%) | 28 (16%) | 4 (2%) | 0 (0%) |
| ECOG Performance Status 0-1 | 151 (89%) | 163 (95%) | 150 (85%) | 144 (82%) |
| Disease Characteristics | ||||
| Baseline Multiple Myeloma Stage (Durie-Salmon) | ||||
| I | 2% | 2% | 6% | 5% |
| II | 31% | 31% | 28% | 33% |
| III | 67% | 67% | 65% | 63% |
| Baseline Creatinine (mg/dL) | ||||
| Median | 1.0 | 1.0 | 0.9 | 0.9 |
| Min,Max | 0.4, 2.6 | 0.5, 2.4 | 0.3, 2.3 | 0.5, 2.3 |
| B2-microglobulin (mg/L) | ||||
| Median | 3.7 | 3.3 | 3.4 | 3.3 |
| Min,Max | 1.1, 45 | 1.3, 15.2 | 1.0, 14.4 | 1.3, 25.3 |
| Number of Prior Therapies | ||||
| No. of Prior Antimyeloma Therapies | ||||
| 1 | 38% | 37% | 32% | 33% |
| ≥2 | 62% | 63% | 68% | 67% |
| Types of Prior Therapies | ||||
| Stem Cell Transplantation | 60% | 60% | 56% | 54% |
| Thalidomide | 42% | 46% | 30% | 38% |
| Dexamethasone | 80% | 70% | 66% | 69% |
| Bortezomib | 11% | 12% | 5% | 4% |
| Melphalan | 34% | 31% | 56% | 52% |
| Doxorubicin | 55% | 52% | 56% | 57% |
The primary efficacy endpoint in both studies was time to progression (TTP). TTP was defined as the time from randomization to the first occurrence of progressive disease or death due to progressive disease.
Preplanned interim analyses of both studies showed that the combination of REVLIMID® (lenalidomide)/dexamethasone was significantly superior to dexamethasone alone for TTP. The studies were unblinded to allow patients in the placebo/dexamethasone group to receive treatment with the REVLIMID® (lenalidomide)/dexamethasone combination.
Table 3 summarizes TTP and response rates based on the best response assessments for Studies 1 and 2.
| 1 NE, Not estimable due to short follow-up. | ||||
| 2 Hazard Ratio of Revlimid/Dexamethasone to Placebo/Dexamethasone | ||||
| 3 The p-value is based on a one-tailed unstratified log rank test. | ||||
| Study 1 | Study 2 | |||
| REVLIMID/Dex
N=170 | Placebo/Dex
N=171 | REVLIMID/Dex
N=176 | Placebo/Dex
N=175 |
|
| TTP | ||||
| Censored n (%) | 115 (68) | 61 (36) | 133 (76) | 78 (45) |
| Median TTP
in weeks [95% CI] |
37.1 [28, NE1] |
19.9 [16, 22] | NE1 |
20 [19.9, 21.6] |
|
Hazard Ratio2 [95% CI] | 0.356 [0.257, 0.494] | 0.392 [0.274, 0.562] | ||
| Log-rank Test p-value 3 | <0.0001 | <0.0001 | ||
| Response | ||||
| Complete Response (CR) n (%) | 14 (8) | 1 (1) | 14 (8) | 1 (1) |
| Partial Response (RR/PR) n (%) | 76 (44) | 27 (16) | 76 (43) | 33 (19) |
| Overall Response n (%) | 90 (53) | 28 (16) | 90 (51) | 34 (19) |
| p-value | <0.0001 | <0.0001 | ||
| Odds Ratio [95% CI] | 5.5 [3.3, 9.1] | 4.3 [2.7, 7.0] | ||
Figures 1 and 2 depict the Kaplan-Meier estimates of TTP in Studies 1 and 2, respectively.
Figure 1: Kaplan-Meier Estimate of Time to Progression - Study 1
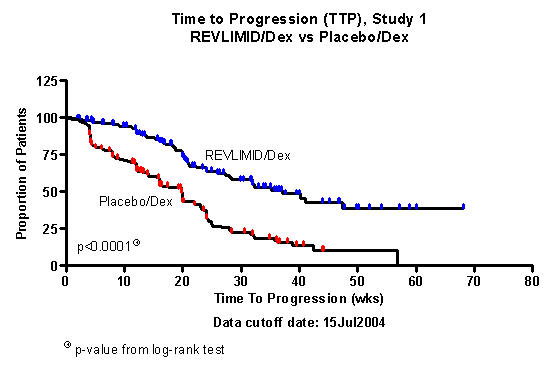
The median duration of Study 1 follow-up was 20.1 weeks.
Figure 2: Kaplan-Meier Estimate of Time to Progression - Study 2
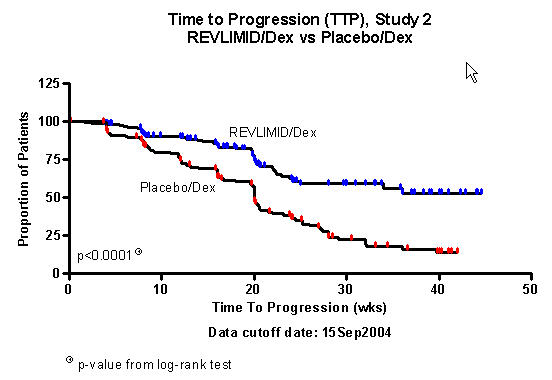
The median duration of Study 2 follow-up was 22.3
weeks.
INDICATIONS AND USAGE
REVLIMID® (lenalidomide) is indicated for the treatment of patients with transfusion-dependent anemia due to Low- or Intermediate-1-risk myelodysplastic syndromes associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalities.
REVLIMID® (lenalidomide) in combination with dexamethasone is indicated for the treatment of multiple myeloma patients who have received at least one prior therapy.
CONTRAINDICATIONS
Pregnancy Category X: (See BOXED WARNINGS)
Due to its structural similarities to thalidomide, a known human teratogen, and data from an embryofetal development study showing treatment with lenalidomide produced malformations in the offspring of female monkeys who received the drug during pregnancy, lenalidomide is contraindicated in pregnant women and women capable of becoming pregnant. (See BOXED WARNINGS.) When there is no alternative, females of childbearing potential may be treated with lenalidomide provided adequate precautions are taken to avoid pregnancy. Females must commit either to abstain continuously from heterosexual sexual intercourse or to use two methods of reliable birth control, including at least one highly effective method (e.g., IUD, hormonal contraception, tubal ligation, or partner’s vasectomy) and one additional effective method (e.g., latex condom, diaphragm, or cervical cap), beginning 4 weeks prior to initiating treatment with REVLIMID® (lenalidomide), during therapy with REVLIMID® (lenalidomide), during therapy delay, and continuing for 4 weeks following discontinuation of REVLIMID® (lenalidomide) therapy. If hormonal or IUD contraception is medically contraindicated, two other effective or highly effective methods may be used.
Females of childbearing potential being treated with REVLIMID® (lenalidomide) should have pregnancy testing (sensitivity of at least 50 mIU/mL). The first test should be performed within 10-14 days and the second test within 24 hours prior to beginning REVLIMID® (lenalidomide) therapy and then weekly during the first month of REVLIMID® (lenalidomide), then monthly thereafter in women with regular menstrual cycles or every 2 weeks in women with irregular menstrual cycles. Pregnancy testing and counseling should be performed if a patient misses her period or if there is any abnormality in menstrual bleeding. If pregnancy occurs, REVLIMID® (lenalidomide) must be immediately discontinued. Under these conditions, the patient should be referred to an obstetrician/gynecologist experienced in reproductive toxicity for further evaluation and counseling.
REVLIMID® (lenalidomide) is contraindicated in any patients who have demonstrated hypersensitivity to the drug or its components.
WARNINGS
Pregnancy Category X: (See BOXED WARNINGS and CONTRAINDICATIONS)
REVLIMID® (lenalidomide) is an analogue of thalidomide. Thalidomide is a known human teratogen that causes life-threatening human birth defects. An embryofetal development study in non-human primates indicates that lenalidomide produced malformations in the offspring of female monkeys who received the drug during pregnancy, similar to birth defects observed in humans following exposure to thalidomide during pregnancy. The teratogenic effect of lenalidomide in humans cannot be ruled out. REVLIMID® (lenalidomide) may cause fetal harm when administered to a pregnant female. Females of childbearing potential should be advised to avoid pregnancy while on REVLIMID® (lenalidomide). Two effective contraceptive methods should be used during therapy, during therapy interruptions and for at least 4 weeks after completing therapy.
There are no adequate and well-controlled studies in pregnant females.
Because of this potential toxicity and to avoid fetal exposure to REVLIMID® (lenalidomide), REVLIMID® (lenalidomide) is only available under a special restricted distribution program. This program is called RevAssist®.
Lenalidomide has been shown to have an embryocidal effect in rabbits at a dose of 50 mg/kg (approximately 120 times the human dose of 10 mg based on body surface area).
An embryo-fetal development study in rats revealed no teratogenic effects at the highest dose of 500 mg/kg (approximately 600 times the human dose of 10 mg based on body surface area). At 100, 300 or 500 mg/kg/day there was minimal maternal toxicity that included slight, transient, reduction in mean body weight gain and food intake. However this animal model may not adequately address the full spectrum of the potential embryo-fetal developmental effects of lenalidomide.
A pre- and post-natal development study in rats revealed few adverse effects on the offspring of female rats treated with lenalidomide at doses up to 500 mg/kg (approximately 600 times the human dose of 10 mg based on body surface area). The male offspring exhibited slightly delayed sexual maturation and the female offspring had slightly lower body weight gains during gestation when bred to male offspring.
The structural similarity of lenalidomide to thalidomide, a known human teratogen, as well as malformations seen in the offspring of female monkeys administered lenalidomide during pregnancy, suggests a potential risk to the developing fetus.
HEMATOLOGIC TOXICITY (NEUTROPENIA AND THROMBOCYTOPENIA):
This drug is associated with significant neutropenia and thrombocytopenia.
Eighty percent of patients with del 5q MDS had to have a dose delay or reduction during the major study for the indication. Thirty-four percent of patients had to have a second dose delay/reduction. Grade 3 or 4 hematologic toxicity was seen in 80% of patients enrolled in the study. In the 48% of patients who developed Grade 3 or 4 neutropenia, the median time to onset was 42 days (range, 14-411 days), and the median time to documented recovery was 17 days (range, 2-170 days). In the 54% of patients who developed Grade 3 or 4 thrombocytopenia, the median time to onset was 28 days (range, 8-290 days), and the median time to documented recovery was 22 days (range, 5-224 days). Patients on therapy for del 5q myelodysplastic syndromes should have their complete blood counts monitored weekly for the first 8 weeks of therapy and at least monthly thereafter. Patients may require dose interruption and/or reduction. Patients may require use of blood product support and/or growth factors. (See DOSAGE AND ADMINISTRATION)
In the pooled multiple myeloma studies Grade 3 and 4 hematologic toxicities were more frequent in patients treated with the combination of REVLIMID® (lenalidomide) and dexamethasone than in patients treated with dexamethasone alone. (See ADVERSE REACTIONS: Table 7.) Patients on therapy should have their complete blood counts monitored every 2 weeks for the first 12 weeks and then monthly thereafter. Patients may require dose interruption and/or dose reduction. (See DOSAGE AND ADMINISTRATION)
DEEP VENOUS THROMBOSIS AND PULMONARY EMBOLISM:
This drug has demonstrated a significantly increased risk of DVT and PE in patients with multiple myeloma who were treated with REVLIMID® (lenalidomide) combination therapy. Patients and physicians are advised to be observant for the signs and symptoms of thromboembolism. Patients should be instructed to seek medical care if they develop symptoms such as shortness of breath, chest pain, or arm or leg swelling. It is not known whether prophylactic anticoagulation or antiplatelet therapy prescribed in conjunction with REVLIMID® (lenalidomide) may lessen the potential for venous thromboembolic events. The decision to take prophylactic measures should be done carefully after an assessment of an individual patient’s underlying risk factors. (See ADVERSE REACTIONS: Table 7)
PRECAUTIONS
Angioedema, Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis
Angioedema and serious dermatologic reactions including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) have been reported. These events can be fatal. Patients with a prior history of Grade 4 rash associated with thalidomide treatment should not receive REVLIMID® (lenalidomide). REVLIMID® (lenalidomide) interruption or discontinuation should be considered for Grade 2-3 skin rash. REVLIMID® (lenalidomide) must be discontinued for angioedema, Grade 4 rash, exfoliative or bullous rash, or if SJS or TEN is suspected, and should not be resumed following discontinuation for these reactions.
Tumor Lysis Syndrome
Lenalidomide has antineoplastic activity and therefore the complications of tumor lysis syndrome may occur. The patients at risk of tumor lysis syndrome are those with high tumor burden prior to treatment. These patients should be monitored closely and appropriate precautions taken.
Information for Patients
Patients should be counseled on lenalidomide’s potential risk of teratogenicity due to its structural similarity to thalidomide and data from an embryofetal development study showing treatment with lenalidomide produced malformations in the offspring of female monkeys who received the drug during pregnancy. Patients may only acquire a prescription for REVLIMID® (lenalidomide) therapy through a controlled distribution program (RevAssist®) through contracted pharmacies. Female patients of childbearing potential will be educated and counseled on the requirements of the RevAssist® program and the precautions to be taken to preclude fetal exposure to REVLIMID® (lenalidomide). Patients should become familiar with the REVLIMID® (lenalidomide) RevAssist® educational materials and Patient Medication Guide, and direct any questions to their physician or pharmacist prior to starting REVLIMID® (lenalidomide) therapy.
Laboratory Tests
The MDS clinical study enrolled patients with absolute neutrophil counts (ANC) ≥ 500/mm3, platelet counts ≥ 50,000/mm3, serum creatinine ≤ 2.5 mg/dL, serum SGOT/AST or SGPT/ALT ≤ 3.0 x upper limit of normal (ULN), and serum direct bilirubin ≤ 2.0 mg/dL. A complete blood cell count (CBC), including white blood cell count with differential, platelet count, hemoglobin, and hematocrit should be performed weekly for the first 8 weeks of REVLIMID® (lenalidomide) treatment and monthly thereafter to monitor for cytopenias.
The multiple myeloma Studies 1 and 2 enrolled patients with absolute neutrophil counts (ANC) ≥ 1000/mm3, platelet counts ≥ 75,000/mm3, serum creatinine ≤ 2.5 mg/dL, serum SGOT/AST or SGPT/ALT ≤ 3.0 x upper limit of normal (ULN), and serum direct bilirubin ≤ 2.0 mg/dL. A CBC should be performed every two weeks for the first three months and at least monthly thereafter to monitor for cytopenias.
Drug Interactions
Results from human in vitro metabolism studies and nonclinical studies show that REVLIMID® (lenalidomide) is neither metabolized by nor inhibits or induces the cytochrome P450 pathway suggesting that lenalidomide is not likely to cause or be subject to P450-based metabolic drug interactions in man.
Co-administration of multiple doses of 10 mg of lenalidomide had no effect on the single dose pharmacokinetics of R- and S-warfarin. Co-administration of single 25-mg dose warfarin had no effect on the pharmacokinetics of total lenalidomide. Expected changes in laboratory assessments of PT and INR were observed after warfarin administration, but these changes were not affected by concomitant lenalidomide administration.
When digoxin was co-administered with lenalidomide the digoxin AUC was not significantly different, however, the digoxin Cmax was increased by 14%. Periodic monitoring of digoxin plasma levels, in accordance with clinical judgment and based on standard clinical practice in patients receiving this medication, is recommended during administration of lenalidomide.
Carcinogenesis, mutagenesis, impairment of fertility
Carcinogenicity: Carcinogenicity studies with lenalidomide have not been conducted.
Mutagenesis: Lenalidomide did not induce mutation in the Ames test, chromosome aberrations in cultured human peripheral blood lymphocytes, or mutation at the thymidine kinase (tk) locus of mouse lymphoma L5178Y cells. Lenalidomide did not increase morphological transformation in Syrian Hamster Embryo assay or induce micronuclei in the polychromatic erythrocytes of the bone marrow of male rats.
Fertility: A fertility and early embryonic development study in rats, with administration of lenalidomide up to 500 mg/kg (approximately 600 times the human dose of 10 mg, based on body surface area) produced no parental toxicity and no adverse effects on fertility.
Pregnancy
Pregnancy Category X: (See BOXED WARNINGS and CONTRAINDICATIONS)
Because of the structural similarity to thalidomide, a known human teratogen, and the data from an embryofetal development study showing treatment with lenalidomide produced malformations in the offspring of female monkeys who received the drug during pregnancy, REVLIMID® (lenalidomide) is contraindicated in females who are or may become pregnant and who are not using the two required types of birth control or who are not continually abstaining from reproductive heterosexual sexual intercourse. REVLIMID® (lenalidomide) should not be used by females who are pregnant or who could become pregnant while taking the drug. If pregnancy does occur during treatment, the drug should be immediately discontinued. Under these conditions, the patient should be referred to an obstetrician/gynecologist experienced in reproductive toxicity for further evaluation and counseling. Any suspected fetal exposure to REVLIMID® (lenalidomide) should be reported to the FDA via the MedWatch program at 1-800-FDA-1088 and also to Celgene Corporation at 1-888-423-5436.
Use in Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for adverse reactions in nursing infants from lenalidomide, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness in pediatric patients below the age of 18 have not been established.
Geriatric Use
REVLIMID® (lenalidomide) has been used in del 5q MDS clinical trials in patients up to 95 years of age.
Of the 148 patients with del 5q MDS enrolled in the major study, 38% were age 65 and over, while 33% were age 75 and over. Although the overall frequency of adverse events (100%) was the same in patients over 65 years of age as in younger patients, the frequency of serious adverse events was higher in patients over 65 years of age than in younger patients (54% vs. 33%). A greater proportion of patients over 65 years of age discontinued from the clinical studies because of adverse events than the proportion of younger patients (27% vs.16%). No differences in efficacy were observed between patients over 65 years of age and younger patients.
REVLIMID® (lenalidomide) has been used in multiple myeloma (MM) clinical trials in patients up to 86 years of age.
Of the 692 MM patients enrolled in Studies 1 and 2, 45% were age 65 or over while 12% of patients were age 75 and over. The percentage of patients age 65 or over was not significantly different between the REVLIMID® (lenalidomide)/dexamethasone and placebo/dexamethasone groups. Of the 346 patients who received REVLIMID® (lenalidomide)/dexamethasone, 46% were age 65 and over. In both studies, patients > 65 years of age were more likely than patients ≤ 65 years of age to experience diarrhea, fatigue, pulmonary embolism, and syncope following use of REVLIMID® (lenalidomide). No differences in efficacy were observed between patients over 65 years of age and younger patients.
Renal Impairment
Since lenalidomide is primarily excreted unchanged by the kidney, adjustments to the starting dose of REVLIMID® (lenalidomide) are recommended to provide appropriate drug exposure in patients with moderate or severe (CLcr < 60 mL/min) renal impairment and in patients on dialysis. See DOSAGE AND ADMINISTRATION.
ADVERSE REACTIONS
Myelodysplastic Syndromes
A total of 148 patients received at least 1 dose of 10 mg lenalidomide in the del 5q MDS clinical study. At least one adverse event was reported in all of the 148 patients who were treated with the 10 mg starting dose of REVLIMID® (lenalidomide). The most frequently reported adverse events were related to blood and lymphatic system disorders, skin and subcutaneous tissue disorders, gastrointestinal disorders, and general disorders and administrative site conditions. (SeePRECAUTIONS)
Thrombocytopenia (61.5%; 91/148) and neutropenia (58.8%; 87/148) were the most frequently reported adverse events observed. The next most common adverse events observed were diarrhea (48.6%; 72/148), pruritus (41.9%; 62/148), rash (35.8%; 53/148) and fatigue (31.1%; 46/148). Table 4 summarizes the adverse events that were reported in ≥ 5% of the REVLIMID® (lenalidomide) treated patients in the del 5q MDS clinical study. Table 5 summarizes the most frequently observed Grade 3 and Grade 4 adverse reactions regardless of relationship to treatment with REVLIMID® (lenalidomide). In the single-arm studies conducted, it is often not possible to distinguish adverse events that are drug-related and those that reflect the patient’s underlying disease.
| NOS, not otherwise specified | ||
| [a] System organ classes and preferred terms are coded using the MedDRA dictionary. System organ classes and preferred terms are listed in descending order of frequency for the Overall column. A patient with multiple occurrences of an AE is counted only once in the AE category. | ||
| System organ class/Preferred term [a] | 10 mg Overall
(N=148) |
|
| Patients with at least one adverse event | 148 | (100.0) |
| Blood and Lymphatic System Disorders | ||
| Thrombocytopenia | 91 | (61.5) |
| Neutropenia | 87 | (58.8) |
| Anemia NOS | 17 | (11.5) |
| Leukopenia NOS | 12 | (8.1) |
| Febrile Neutropenia | 8 | (5.4) |
| Skin and Subcutaneous Tissue Disorders | ||
| Pruritus | 62 | (41.9) |
| Rash NOS | 53 | (35.8) |
| Dry Skin | 21 | (14.2) |
| Contusion | 12 | (8.1) |
| Night Sweats | 12 | (8.1) |
| Sweating Increased | 10 | (6.8) |
| Ecchymosis | 8 | (5.4) |
| Erythema | 8 | (5.4) |
| Gastrointestinal Disorders | ||
| Diarrhea NOS | 72 | (48.6) |
| Constipation | 35 | (23.6) |
| Nausea | 35 | (23.6) |
| Abdominal Pain NOS | 18 | (12.2) |
| Vomiting NOS | 15 | (10.1) |
| Abdominal Pain Upper | 12 | (8.1) |
| Dry Mouth | 10 | (6.8) |
| Loose Stools | 9 | (6.1) |
| Respiratory, Thoracic and Mediastinal Disorders | ||
| Nasopharyngitis | 34 | (23.0) |
| Cough | 29 | (19.6) |
| Dyspnea NOS | 25 | (16.9) |
| Pharyngitis | 23 | (15.5) |
| Epistaxis | 22 | (14.9) |
| Dyspnea Exertional | 10 | (6.8) |
| Rhinitis NOS | 10 | (6.8) |
| Bronchitis NOS | 9 | (6.1) |
| General Disorders and Administration Site Conditions | ||
| Fatigue | 46 | (31.1) |
| Pyrexia | 31 | (20.9) |
| Edema Peripheral | 30 | (20.3) |
| Asthenia | 22 | (14.9) |
| Edema NOS | 15 | (10.1) |
| Pain NOS | 10 | (6.8) |
| Rigors | 9 | (6.1) |
| Chest Pain | 8 | (5.4) |
| Musculoskeletal and Connective Tissue Disorders | ||
| Arthralgia | 32 | (21.6) |
| Back Pain | 31 | (20.9) |
| Muscle Cramp | 27 | (18.2) |
| Pain in Limb | 16 | (10.8) |
| Myalgia | 13 | (8.8) |
| Peripheral Swelling | 12 | (8.1) |
| Nervous System Disorders | ||
| Dizziness | 29 | (19.6) |
| Headache | 29 | (19.6) |
| Hypoesthesia | 10 | (6.8) |
| Dysgeusia | 9 | (6.1) |
| Peripheral Neuropathy NOS | 8 | (5.4) |
| Infections and Infestations | ||
| Upper Respiratory Tract Infection NOS | 22 | (14.9) |
| Pneumonia NOS | 17 | (11.5) |
| Urinary Tract Infection NOS | 16 | (10.8) |
| Sinusitis NOS | 12 | (8.1) |
| Cellulitis | 8 | (5.4) |
| Metabolism and Nutrition Disorders | ||
| Hypokalemia | 16 | (10.8) |
| Anorexia | 15 | (10.1) |
| Hypomagnesemia | 9 | (6.1) |
| Investigations | ||
| Alanine Aminotransferase Increased | 12 | (8.1) |
| Psychiatric Disorders | ||
| Insomnia | 15 | (10.1) |
| Depression | 8 | (5.4) |
| Vascular Disorders | ||
| Hypertension NOS | 9 | (6.1) |
| Renal and Urinary Disorders | ||
| Dysuria | 10 | (6.8) |
| Cardiac Disorders | ||
| Palpitations | 8 | (5.4) |
| Endocrine Disorders | ||
| Acquired Hypothyroidism | 10 | (6.8) |
| [1] Adverse events with frequency ≥1% in the 10 mg Overall group. Grade 3 and 4 are based on National Cancer Institute Common Toxicity Criteria version 2. | ||
| [2] Preferred Terms are coded using the MedDRA dictionary. A patient with multiple occurrences of an AE is counted only once in the Preferred Term category. | ||
| Preferred term [2] | 10 mg
(N=148) |
|
| Patients with at least one Grade 3/4 AE | 131 | (88.5) |
| Neutropenia | 79 | (53.4) |
| Thrombocytopenia | 74 | (50.0) |
| Pneumonia NOS | 11 | (7.4) |
| Rash NOS | 10 | (6.8) |
| Anemia NOS | 9 | (6.1) |
| Leukopenia NOS | 8 | (5.4) |
| Fatigue | 7 | (4.7) |
| Dyspnea | 7 | (4.7) |
| Back Pain | 7 | (4.7) |
| Febrile Neutropenia | 6 | (4.1) |
| Nausea | 6 | (4.1) |
| Diarrhea NOS | 5 | (3.4) |
| Pyrexia | 5 | (3.4) |
| Sepsis | 4 | (2.7) |
| Dizziness | 4 | (2.7) |
| Granulocytopenia | 3 | (2.0) |
| Chest Pain | 3 | (2.0) |
| Pulmonary Embolism | 3 | (2.0) |
| Respiratory Distress | 3 | (2.0) |
| Pruritus | 3 | (2.0) |
| Pancytopenia | 3 | (2.0) |
| Muscle Cramp | 3 | (2.0) |
| Respiratory Tract Infection | 2 | (1.4) |
| Upper Respiratory Tract Infection | 2 | (1.4) |
| Asthenia | 2 | (1.4) |
| Multi-organ Failure | 2 | (1.4) |
| Epistaxis | 2 | (1.4) |
| Hypoxia | 2 | (1.4) |
| Pleural Effusion | 2 | (1.4) |
| Pneumonitis NOS | 2 | (1.4) |
| Pulmonary Hypertension NOS | 2 | (1.4) |
| Vomiting NOS | 2 | (1.4) |
| Sweating Increased | 2 | (1.4) |
| Arthralgia | 2 | (1.4) |
| Pain in Limb | 2 | (1.4) |
| Headache | 2 | (1.4) |
| Syncope | 2 | (1.4) |
In other clinical studies of REVLIMID® (lenalidomide) in MDS patients, the following serious adverse events (regardless of relationship to study drug treatment) not described in Table 4 or 5 were reported:
Blood and lymphatic system disorders: warm type hemolytic anemia, splenic infarction, bone marrow depression NOS, coagulopathy, hemolysis NOS, hemolytic anemia NOS, refractory anemia
Cardiac disorders: cardiac failure congestive, atrial fibrillation, angina pectoris, cardiac arrest, cardiac failure NOS, cardio-respiratory arrest, cardiomyopathy NOS, myocardial infarction, myocardial ischemia, atrial fibrillation aggravated, bradycardia NOS, cardiogenic shock, pulmonary edema NOS, supraventricular arrhythmia NOS, tachyarrhythmia, ventricular dysfunction
Ear and labyrinth disorders: vertigo
Endocrine disorders: Basedow’s disease
Gastrointestinal disorders: gastrointestinal hemorrhage NOS, colitis ischemic, intestinal perforation NOS, rectal hemorrhage, colonic polyp, diverticulitis NOS, dysphagia, gastritis NOS, gastroenteritis NOS, gastroesophageal reflux disease, obstructive inguinal hernia, irritable bowel syndrome, melena, pancreatitis due to biliary obstruction, pancreatitis NOS, perirectal abscess, small intestinal obstruction NOS, upper gastrointestinal hemorrhage
General disorders and administration site conditions: disease progression NOS, fall, gait abnormal, intermittent pyrexia, nodule, rigors, sudden death
Hepatobiliary disorders: hyperbilirubinemia, cholecystitis acute NOS, cholecystitis NOS, hepatic failure
Immune system disorders: hypersensitivity NOS
Infections and infestations: infection NOS, bacteremia, central line infection, clostridial infection NOS, ear infection NOS, Enterobacter sepsis, fungal infection NOS, herpes viral infection NOS, influenza, kidney infection NOS, Klebsiella sepsis, lobar pneumonia NOS, localized infection, oral infection, Pseudomonas infection NOS, septic shock, sinusitis acute NOS, sinusitis NOS, Staphylococcal infection, urosepsis
Injury, poisoning and procedural complications: femur fracture, transfusion reaction, cervical vertebral fracture, femoral neck fracture, fractured pelvis NOS, hip fracture, overdose NOS, post procedural hemorrhage, rib fracture, road traffic accident, spinal compression fracture
Investigations: blood creatinine increased, culture NOS negative, hemoglobin decreased, liver function tests NOS abnormal, troponin I increased
Metabolism and nutrition disorders: dehydration, gout, hypernatremia, hypoglycemia NOS
Musculoskeletal and connective tissue disorders: arthritis NOS, arthritis NOS aggravated, gouty arthritis, neck pain, chondrocalcinosis pyrophosphate
Neoplasms benign, malignant and unspecified: acute leukemia NOS, acute myeloid leukemia NOS, bronchoalveolar carcinoma, lung cancer metastatic, lymphoma NOS, prostate cancer metastatic
Nervous system disorders: cerebrovascular accident, aphasia, cerebellar infarction, cerebral infarction, depressed level of consciousness, dysarthria, migraine NOS, spinal cord compression NOS, subarachnoid hemorrhage NOS, transient ischemic attack
Psychiatric disorders: confusional state
Renal and urinary disorders: renal failure NOS, hematuria, renal failure acute, azotemia, calculus ureteric, renal mass NOS
Reproductive system and breast disorders: pelvic pain NOS
Respiratory, thoracic and mediastinal disorders: bronchitis NOS, chronic obstructive airways disease exacerbated, respiratory failure, dyspnea exacerbated, interstitial lung disease, lung infiltration NOS, wheezing
Skin and subcutaneous tissue disorders: acute febrile neutrophilic dermatosis
Vascular system disorders: deep vein thrombosis, hypotension NOS, aortic disorder, ischemia NOS, thrombophlebitis superficial, thrombosis
Multiple Myeloma
Data were evaluated from 691 patients in two studies who received at least one dose of REVLIMID® (lenalidomide)/dexamethasone (346 patients) or placebo/dexamethasone (345 patients).
In the REVLIMID® (lenalidomide)/dexamethasone treatment group, 151 patients (45%) underwent at least one dose interruption with or without a dose reduction of REVLIMID® (lenalidomide) compared to 21% in the placebo/dexamethasone treatment group. Of these patients who had one dose interruption with or without a dose reduction, 50% in the REVLIMID® (lenalidomide)/dexamethasone treatment group underwent at least one additional dose interruption with or without a dose reduction compared to 21% in the placebo/dexamethasone treatment group. Most adverse events and Grade 3/4 adverse events were more frequent in patients who received the combination of REVLIMID® (lenalidomide)/dexamethasone compared to placebo/dexamethasone.
Table 6 summarizes the number and percentage of patients with Grade 1-4 adverse events reported in ≥10% of patients in either treatment group in Studies 1 and 2.
| ª See WARNINGS | ||||
| Revlimid/Dex (N=346) | Placebo/Dex (N=345) |
|||
| System organ class/Preferred term | n | (%) | n | (%) |
| Subjects with at least one adverse event | 346 | (100.0) | 344 | (99.7) |
| Blood and Lymphatic System Disorders | ||||
| Neutropenia | 96 | (27.7) | 16 | (4.6) |
| Anemia NOS | 84 | (24.3) | 60 | (17.4) |
| Thrombocytopenia | 59 | (17.1) | 34 | (9.9) |
| Eye Disorders | ||||
| Vision Blurred | 51 | (14.7) | 36 | (10.4) |
| Gastrointestinal Disorders | ||||
| Constipation | 134 | (38.7) | 64 | (18.6) |
| Diarrhea NOS | 101 | (29.2) | 85 | (24.6) |
| Nausea | 76 | (22.0) | 66 | (19.1) |
| Dyspepsia | 48 | (13.9) | 46 | (13.3) |
| Vomiting NOS | 35 | (10.1) | 28 | (8.1) |
| General Disorders and Administration Site Conditions | ||||
| Fatigue | 133 | (38.4) | 129 | (37.4) |
| Asthenia | 81 | (23.4) | 86 | (24.9) |
| Pyrexia | 80 | (23.1) | 67 | (19.4) |
| Edema Peripheral | 73 | (21.1) | 65 | (18.8) |
| Infections and Infestations | ||||
| Upper Respiratory Tract Infection NOS | 47 | (13.6) | 43 | (12.5) |
| Pneumonia NOS | 39 | (11.3) | 26 | (7.5) |
| Investigations | ||||
| Weight Decreased | 63 | (18.2) | 48 | (13.9) |
| Metabolism and Nutrition Disorders | ||||
| Hyperglycemia NOS | 52 | (15.0) | 49 | (14.2) |
| Anorexia | 47 | (13.6) | 30 | (8.7) |
| Hypokalemia | 39 | (11.3) | 18 | (5.2) |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Muscle Cramp | 104 | (30.1) | 71 | (20.6) |
| Back Pain | 53 | (15.3) | 49 | (14.2) |
| Muscle Weakness NOS | 52 | (15.0) | 53 | (15.4) |
| Arthralgia | 36 | (10.4) | 51 | (14.8) |
| Nervous System Disorders | ||||
| Headache | 74 | (21.4) | 74 | (21.4) |
| Dizziness | 72 | (20.8) | 53 | (15.4) |
| Tremor | 68 | (19.7) | 24 | (7.0) |
| Dysgeusia | 46 | (13.3) | 32 | (9.3) |
| Paresthesia | 40 | (11.6) | 43 | (12.5) |
| Psychiatric Disorders | ||||
| Insomnia | 111 | (32.1) | 128 | (37.1) |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Dyspnea NOS | 70 | (20.2) | 53 | (15.4) |
| Cough | 50 | (14.5) | 71 | (20.6) |
| Skin and Subcutaneous Tissue Disorders | ||||
| Rash NOS | 55 | (15.9) | 28 | (8.1) |
| Vascular Disorders | ||||
| Deep Vein Thrombosisa | 27 | (7.8) | 11 | (3.2) |
| Pulmonary Embolisma | 11 | (3.2) | 3 | (0.9) |
Table 7 summarizes the Grade 3/4 adverse events reported in ≥2% of patients in either treatment group in Studies 1 and 2.
| ª See WARNINGS | ||||||||
| Revlimid/Dex (N=346) | Placebo/Dex (N=345) | |||||||
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | |||||
| System organ class/ Preferred term | n | (%) | n | (%) | n | (%) | n | (%) |
| Patients with at least one Grade 3 or 4 AE | 225 | (65.0) | 25 | (7.2) | 186 | (53.9) | 31 | (9.0) |
| Blood and Lymphatic System Disorders | ||||||||
| Neutropenia | 60 | (17.3) | 13 | (3.8) | 8 | (2.3) | 2 | (0.6) |
| Thrombocytopenia | 31 | (9.0) | 4 | (1.2) | 16 | (4.6) | 3 | (0.9) |
| Anemia NOS | 25 | (7.2) | 4 | (1.2) | 10 | (2.9) | 2 | (0.6) |
| Leukopenia NOS | 12 | (3.5) | 0 | (0.0) | 1 | (0.3) | 0 | (0.0) |
| Lymphopenia | 8 | (2.3) | 0 | (0.0) | 4 | (1.2) | 0 | (0.0) |
| Cardiac Disorders | ||||||||
| Atrial Fibrillation | 9 | (2.6) | 1 | (0.3) | 2 | (0.6) | 1 | (0.3) |
| Gastrointestinal Disorders | ||||||||
| Diarrhea NOS | 8 | (2.3) | 0 | (0.0) | 2 | (0.6) | 0 | (0.0) |
| Constipation | 7 | (2.0) | 0 | (0.0) | 1 | (0.3) | 0 | (0.0) |
| General Disorders and Administration Site Conditions | ||||||||
| Fatigue | 20 | (5.8) | 1 | (0.3) | 13 | (3.8) | 0 | (0.0) |
| Asthenia | 14 | (4.0) | 0 | (0.0) | 16 | (4.6) | 0 | (0.0) |
| Pyrexia | 4 | (1.2) | 0 | (0.0) | 8 | (2.3) | 0 | (0.0) |
| Infections and Infestations | ||||||||
| Pneumonia NOS | 18 | (5.2) | 4 | (1.2) | 15 | (4.3) | 3 | (0.9) |
| Metabolism and Nutrition Disorders | ||||||||
| Hyperglycemia NOS | 22 | (6.4) | 4 | (1.2) | 19 | (5.5) | 7 | (2.0) |
| Hypocalcemia | 8 | (2.3) | 5 | (1.4) | 4 | (1.2) | 1 | (0.3) |
| Hypokalemia | 9 | (2.6) | 1 | (0.3) | 5 | (1.4) | 0 | (0.0) |
| Musculoskeletal and Connective Tissue Disorders | ||||||||
| Muscle Weakness NOS | 18 | (5.2) | 0 | (0.0) | 10 | (2.9) | 0 | (0.0) |
| Nervous System Disorders | ||||||||
| Syncope | 7 | (2.0) | 0 | (0.0) | 3 | (0.9) | 0 | (0.0) |
| Neuropathy NOS | 7 | (2.0) | 0 | (0.0) | 2 | (0.6) | 0 | (0.0) |
| Psychiatric Disorders | ||||||||
| Depression | 9 | (2.6) | 0 | (0.0) | 5 | (1.4) | 1 | (0.3) |
| Confusional State | 6 | (1.7) | 0 | (0.0) | 8 | (2.3) | 0 | (0.0) |
| Respiratory, Thoracic and Mediastinal Disorders | ||||||||
| Dyspnea NOS | 6 | (1.7) | 3 | (0.9) | 7 | (2.0) | 1 | (0.3) |
| Vascular Disorders | ||||||||
| Deep Vein Thrombosisa | 23 | (6.6) | 1 | (0.3) | 9 | (2.6) | 1 | (0.3) |
| Pulmonary Embolisma | 2 | (0.6) | 9 | (2.6) | 1 | (0.3) | 2 | (0.6) |
Thrombotic Events (See WARNINGS)
In the pooled analysis, thrombotic or thromboembolic events, including deep vein thrombosis, pulmonary embolism, thrombosis, and intracranial venous sinus thrombosis, were reported more frequently in patients treated with REVLIMID® (lenalidomide)/dexamethasone combination. The number of patients experiencing a thrombotic event in the combination arm were 43/346 (12%) compared with those in the placebo/dexamethasone arm 14/345 (4%).
In these and other clinical studies of REVLIMID® (lenalidomide) in patients with multiple myeloma, the following serious adverse events (considered related to study drug treatment) not described in Table 7 were reported:
Blood and lymphatic system disorders: pancytopenia, anemia NOS aggravated
Cardiac disorders: cardiac failure congestive, atrial flutter, pulmonary edema
Endocrine disorders: adrenal insufficiency NOS, acquired hypothyroidism
Eye disorders: blindness
Gastrointestinal disorders: abdominal pain NOS, colitis pseudomembranous, gastritis NOS, gastrointestinal hemorrhage NOS, peptic ulcer hemorrhage, upper gastrointestinal hemorrhage
General disorders and administration site conditions: performance status decreased
Hepatobiliary disorders: hepatic failure, hepatitis toxic
Infections and infestations: bronchopneumonia NOS, cellulitis, Pneumocystis carinii pneumonia, sepsis NOS, bursitis infective NOS, cellulitis staphylococcal, Enterobacter bacteremia, Escherichia sepsis, gastrointestinal infection NOS, herpes zoster, herpes zoster ophthalmic, infection NOS, lung infection NOS, neutropenic sepsis, pneumonia bacterial NOS, pneumonia cytomegaloviral, pneumonia pneumococcal, pneumonia primary atypical, pneumonia staphylococcal, septic shock, streptococcal sepsis, subacute endocarditis, urinary tract infection NOS
Investigations: International normalized ratio increased, weight decreased, blood creatinine increased, body temperature increased, c-reactive protein increased, hemoglobin decreased, white blood cell count decreased
Metabolism and nutrition disorders: dehydration, diabetes mellitus NOS, diabetes with hyperosmolarity, diabetic ketoacidosis
Musculoskeletal and connective tissue disorders: myopathy steroid, back pain, myopathy
Nervous system disorders: dizziness, memory impairment, brain edema, cerebral infarction, cerebral ischemia, cerebrovascular accident, encephalitis NOS, intracranial hemorrhage NOS, intracranial venous sinus thrombosis NOS, leukoencephalopathy, somnolence, tremor
Psychiatric disorders: mental status changes, delirium, delusion NOS, insomnia, psychotic disorder NOS
Renal and urinary disorders: Fanconi syndrome acquired, hematuria, renal failure acute, renal failure NOS, renal tubular necrosis, urinary retention
Respiratory, thoracic and mediastinal disorders: bronchopneumopathy, hypoxia
Skin and subcutaneous tissue disorders: rash NOS, skin desquamation NOS
Vascular system disorders: phlebitis NOS, venous thrombosis NOS limb, circulatory collapse, hypertension NOS, hypotension NOS, orthostatic hypotension, peripheral ischemia
OVERDOSAGE
No cases of overdose have been reported during the clinical studies.
DOSAGE AND ADMINISTRATION
Myelodysplastic Syndromes
The recommended starting dose of REVLIMID® (lenalidomide) is 10 mg daily with water. Patients should not break, chew or open the capsules. Dosing is continued or modified based upon clinical and laboratory findings.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it would be prudent to monitor renal function.
Dose Adjustments During Treatment
Patients who are dosed initially at 10 mg and who experience thrombocytopenia should have their dosage adjusted as follows:
Platelet counts
| If baseline ≥100,000/mcL | |
| When Platelets | Recommended Course |
| Fall to <50,000/mcL | Interrupt REVLIMID® treatment |
| Return to ≥50,000/mcL | Resume REVLIMID® at 5 mg daily |
| If baseline <100,000/mcL | |
| When Platelets | Recommended Course |
| Fall to 50% of the baseline value | Interrupt REVLIMID® treatment |
| If baseline
≥60,000/mcL and returns to ≥50,000/mcL | Resume REVLIMID® at 5 mg daily |
| If baseline
<60,000/mcL and returns to ≥30,000/mcL | Resume REVLIMID® at 5 mg daily |
| When Platelets | Recommended Course |
| <30,000/mcL or
<50,000/mcL and platelet transfusions | Interrupt REVLIMID® treatment |
| Return to
≥30,000/mcL (without hemostatic failure) | Resume REVLIMID® at 5 mg daily |
Patients who experience thrombocytopenia at 5 mg daily should have their dosage adjusted as follows:
| When Platelets | Recommended Course |
| <30,000/mcL or
<50,000/mcL and platelet transfusions | Interrupt REVLIMID® treatment |
| Return to
≥30,000/mcL (without hemostatic failure) | Resume REVLIMID® at 5 mg every other day |
Patients who are dosed initially at 10 mg and experience neutropenia should have their dosage adjusted as follows:
Neutrophil counts (ANC)+
| If baseline ANC ≥1,000/mcL | |
| When Neutrophils | Recommended Course |
| Fall to <750/mcL | Interrupt REVLIMID® treatment |
| Return to ≥1,000/mcL | Resume REVLIMID® at 5 mg daily |
| If baseline ANC <1,000/mcL | |
| When Neutrophils | Recommended Course |
| Fall to <500/mcL | Interrupt REVLIMID® treatment |
| Return to ≥500/mcL | Resume REVLIMID® at 5 mg daily |
| When Neutrophils | Recommended Course |
| <500/mcL for
≥7 days or
<500/mcL associated with fever (≥38.5°C) | Interrupt REVLIMID® treatment |
| Return to ≥500/mcL | Resume REVLIMID® at 5 mg daily |
Patients who experience neutropenia at 5 mg daily should have their dosage adjusted as follows:
| + Absolute neutrophil count | |
| When Neutrophils | Recommended Course |
| <500/mcL for
≥7 days or
<500/mcL associated with fever (≥38.5°C) | Interrupt REVLIMID® treatment |
| Return to ≥500/mcL | Resume REVLIMID® at 5 mg every other day |
Multiple Myeloma
The recommended starting dose of REVLIMID® (lenalidomide) is 25 mg/day with water orally administered as a single 25 mg capsule on Days 1‑21 of repeated 28-day cycles. Patients should not break, chew or open the capsules. The recommended dose of dexamethasone is 40 mg/day on Days 1‑4, 9‑12, and 17‑20 of each 28‑day cycle for the first 4 cycles of therapy and then 40 mg/day orally on Days 1‑4 every 28 days. Dosing is continued or modified based upon clinical and laboratory findings.
The effect of substituting lesser strengths of REVLIMID® (lenalidomide) to achieve a 25 mg capsule dose is unknown.
Dose Adjustments During Treatment
Dose modification guidelines, as summarized below are recommended to manage Grade 3 or 4 neutropenia or thrombocytopenia or other Grade 3 or 4 toxicity judged to be related to lenalidomide.
Platelet counts
| When Platelets | Recommended Course |
| Fall to <30,000/mcL | Interrupt REVLIMID® treatment, follow CBC weekly |
| Return to ≥30,000/mcL | Restart REVLIMID® at 15 mg daily |
| For each subsequent drop <30,000/mcL | Interrupt REVLIMID® treatment |
| Return to ≥30,000/mcL | Resume REVLIMID® at 5 mg less than the previous dose. Do not dose below 5 mg daily |
Neutrophil counts (ANC)
| When Neutrophils | Recommended Course |
| Fall to <1000/mcL | Interrupt REVLIMID® treatment, add G-CSF, follow CBC weekly |
| Return to ≥1,000/mcL and neutropenia is the only toxicity | Resume REVLIMID® at 25 mg daily |
| Return to
≥1,000/mcL and if other toxicity | Resume REVLIMID® at 15 mg daily |
| For each subsequent drop <1,000/mcL | Interrupt REVLIMID® treatment |
| Return to ≥1,000/mcL | Resume REVLIMID® at 5 mg less than the previous dose. Do not dose below 5 mg daily |
Starting Dose Adjustment for Renal Impairment:
Since lenalidomide is primarily excreted unchanged by the kidney, adjustments to the starting dose of REVLIMID® (lenalidomide) are recommended to provide appropriate drug exposure in patients with moderate or severe renal impairment and in patients on dialysis. Based on a pharmacokinetic study in patients with renal impairment due to nonmalignant conditions, lenalidomide starting dose adjustment is recommended for patients with CLcr < 60 mL/min. Non-dialysis patients with creatinine clearances less than 11 mL/min, and dialysis patients with creatinine clearances less than 7 mL/min, have not been studied. The recommendations for initial starting doses for patients with multiple myeloma (MM) and myelodysplastic syndromes (MDS) are as follows:
| Category | Renal Function (Cockcroft-Gault CLcr) | Disease | |
| Multiple Myeloma | Myelodysplastic Syndromes | ||
| Moderate Renal Impairment | 30 ≤ CLcr < 60 mL/min | 10 mg Every 24 hours | 5 mg Every 24 hours |
| Severe Renal Impairment | CLcr < 30 ml/min (not requiring dialysis) | 15 mg Every 48 hours | 5 mg Every 48 hours |
| End Stage Renal Disease | CLcr < 30 mL/min (requiring dialysis) | 5 mg Once daily. On dialysis days the dose should be administered following dialysis | 5 mg 3 times a week following each dialysis |
After initiation of REVLIMID® (lenalidomide) therapy, subsequent REVLIMID® (lenalidomide) dose modification should be based on individual patient treatment tolerance, as described elsewhere in this section.
Other Grade 3/4 Toxicities
For other Grade 3/4 toxicities judged to be related to lenalidomide, hold treatment and restart at next lower dose level when toxicity has resolved to ≤ Grade 2.
HOW SUPPLIED
REVLIMID® (lenalidomide) 5 mg, 10 mg, 15 mg and 25 mg capsules will be supplied through the RevAssist® program. (See INFORMATION FOR PATIENTS)
REVLIMID® (lenalidomide) is supplied as:
White opaque capsules imprinted “REV” on one half and “5 mg” on the other half in black ink:
5 mg bottles of 28 (NDC 59572-405-28)
5 mg bottles of 100 (NDC 59572-405-00)
Blue/green and pale yellow opaque capsules imprinted “REV” on one half and “10 mg” on the other half in black ink:
10 mg bottles of 28 (NDC 59572-410-28)
10 mg bottles of 100 (NDC 59572-410-00)
Powder blue and white opaque capsules imprinted “REV” on one half and “15 mg” on the other half in black ink:
15 mg bottles of 21 (NDC 59572-415-21)
15 mg bottles of 100 (NDC 59572-415-00)
White opaque capsules imprinted “REV” on one half and “25 mg” on the other half in black ink:
25 mg bottles of 21 (NDC 59572-425-21)
25 mg bottles of 100 (NDC 59572-425-00)
Storage and Dispensing
Dispense no more than a 28-day supply.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F). [See USP Controlled Room Temperature].
Rx only.
Manufactured for Celgene Corporation
86 Morris Avenue
Summit, NJ 07901
Important Information and WARNINGS for All Patients Taking REVLIMID® (lenalidomide)
WARNING: POTENTIAL FOR HUMAN BIRTH DEFECTS.
LENALIDOMIDE IS AN ANALOGUE OF THALIDOMIDE. THALIDOMIDE IS A KNOWN HUMAN TERATOGEN THAT CAUSES LIFE-THREATENING HUMAN BIRTH DEFECTS. IF LENALIDOMIDE IS TAKEN DURING PREGNANCY, IT MAY CAUSE BIRTH DEFECTS OR DEATH TO AN UNBORN BABY. FEMALES SHOULD BE ADVISED TO AVOID PREGNANCY WHILE ON LENALIDOMIDE.
All Patients
- The patient understands that birth defects may occur with the use of REVLIMID® (lenalidomide).
- The patient has been warned by his/her doctor that an unborn baby may have birth defects and can even die, if a female is pregnant or becomes pregnant while taking REVLIMID® (lenalidomide).
- REVLIMID® (lenalidomide) will be prescribed ONLY for the patient and must NOT be shared with ANYONE, even someone who has similar symptoms.
- REVLIMID® (lenalidomide) must be kept out of the reach of children and should NEVER be given to females who are able to have children.
- The patient cannot donate blood while taking REVLIMID® (lenalidomide).
- The patient has read the REVLIMID® (lenalidomide) patient brochure and understands the contents, including other possible health problems from REVLIMID® (lenalidomide), “side effects.”
- The patient’s doctor has answered any questions the patient has asked.
- The patient must participate in a telephone survey and patient registry, while taking REVLIMID® (lenalidomide).
Female Patients of Childbearing Potential
- The patient must not take REVLIMID® (lenalidomide) if she is pregnant, breast-feeding a baby, or able to get pregnant and not using the required two methods of birth control.
- The patient confirms that she is not now pregnant, nor will she try to become pregnant during REVLIMID® (lenalidomide) therapy, during therapy interruption and for at least 4 weeks after she has completely finished taking REVLIMID® (lenalidomide).
-
If the patient is able to become pregnant, she must use at least one highly effective method and one additional effective method of birth control (contraception) AT THE SAME TIME:
At least one highly effective method AND One additional effective method IUD Latex condom Hormonal (birth control pills,
injections, patch or implants)Diaphragm Tubal ligation Cervical cap Partner's vasectomy - These birth control methods must be used for at least 4 weeks before beginning REVLIMID® (lenalidomide) therapy, during REVLIMID® (lenalidomide) therapy, during therapy interruption and for 4 weeks following discontinuation of REVLIMID® (lenalidomide) therapy.
- The patient must use these birth control methods unless she completely abstains from heterosexual sexual contact.
- If a hormonal method (birth control pills, injections, patch or implants) or IUD is not medically possible for the patient, she may use another highly effective method or two barrier methods AT THE SAME TIME.
- The patient must have a pregnancy test done by her doctor within 10-14 days and 24 hours before REVLIMID® (lenalidomide) therapy, then weekly during the first 4 weeks of REVLIMID® (lenalidomide) therapy.
- Thereafter, the patient must have a pregnancy test every 4 weeks if she has regular menstrual cycles, or every 2 weeks if her cycles are irregular while she is taking REVLIMID® (lenalidomide).
- The patient must immediately stop taking
REVLIMID® (lenalidomide) and
inform her doctor:
- If she becomes pregnant while taking the drug.
- If she misses her menstrual period, or experiences unusual menstrual bleeding.
- If she stops using birth control.
- If she thinks FOR ANY REASON that she may be pregnant.
- The patient understands that if her doctor is not available, she can call 1-888-668-2528 for information on emergency contraception.
Female Patients Not of Childbearing Potential
- The patient certifies that she is not now pregnant, nor of childbearing potential as she has been postmenopausal naturally for at least 24 months (been through the change of life); or she has had a hysterectomy or bilateral oophorectomy.
- The patient or guardian certifies that a prepubertal female child is not now pregnant, nor is of childbearing potential as menstruation has not yet begun, and/or the child will not be engaging in heterosexual sexual contact for at least 4 weeks before REVLIMID® (lenalidomide) therapy, during REVLIMID®(lenalidomide) therapy, during therapy interruption and for at least 4 weeks after stopping therapy.
Male Patients
- The patient has been told by his doctor that he must NEVER have unprotected sexual contact with a female who can become pregnant.
- Because it is not known whether REVLIMID® (lenalidomide) is present in semen, his doctor has explained that he must either completely abstain from sexual contact with females who are pregnant or able to become pregnant, or he must use a latex condom EVERY TIME he engages in any sexual contact with females who are pregnant or may become pregnant while he is taking REVLIMID® (lenalidomide) and for 4 weeks after he stops taking the drug, even if he has had a successful vasectomy.
- The patient should inform his doctor:
- If he has had unprotected sexual contact with a female who can become pregnant.
- If he thinks FOR ANY REASON, that his sexual partner may be pregnant.
- The patient understands that if his doctor is not available, he can call 1-888-668-2528 for information on emergency contraception.
- The patient cannot donate semen or sperm while taking REVLIMID® (lenalidomide).
RevPlyPI.006/MG.006 01/09
Information for patients and caregivers:
MEDICATION GUIDE
REVLIMID® (rev-li-mid)
(lenalidomide)
Read the Medication Guide that comes with REVLIMID® before you start taking it and each time you get a new prescription. There may be new information. This Medication Guide does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about REVLIMID®?
- REVLIMID® is only for patients who understand and agree to all of the instructions in the RevAssist® program.
-
REVLIMID® may
cause serious side effects including:
- birth defects
- low white blood cells and platelets
- blood clots in veins and in the lungs
-
Possible birth defects (deformed
babies) or death of an unborn baby. Female
patients who are pregnant or who plan to become pregnant
must not take
REVLIMID®.
REVLIMID® is similar to the medicine thalidomide (THALOMID®). We know thalidomide causes life-threatening birth defects. REVLIMID® has not been tested in pregnant women. REVLIMID® has harmed unborn animals in animal testing.
Female patients must not get pregnant:
- for 4 weeks before starting REVLIMID®
- while taking REVLIMID®
- during dose interruptions of REVLIMID®
- for 4 weeks after stopping REVLIMID®
It is not known if REVLIMID® passes into semen, so:
- Male patients, including those who have had a vasectomy, must use a latex condom during any sexual contact with a pregnant female or a female that can become pregnant while taking REVLIMID® and for 4 weeks after stopping REVLIMID®.
- FDA MedWatch at 1-800-FDA-1088, and
- Celgene Corporation at 1-888-423-5436
- Low white blood cells (neutropenia) and low platelets (thrombocytopenia). REVLIMID® causes low white blood cells and low platelets in most patients. You may need a blood transfusion or certain medicines if your blood counts drop too low. If you are being treated for del 5q myelodysplastic syndromes (MDS) your blood counts should be checked weekly during the first 8 weeks of treatment with REVLIMID®, and at least monthly thereafter. If you are being treated for multiple myeloma, your blood counts should be checked every 2 weeks for the first 12 weeks and then at least monthly thereafter.
-
An increased chance for blood
clots in veins and in the lungs. Call your
healthcare provider or get emergency medical care right away
if you get the following signs or symptoms:
- shortness of breath
- chest pain
- arm or leg swelling
What is REVLIMID® and what is it used for?
REVLIMID® is a medicine taken by mouth to treat certain patients who have myelodysplastic syndromes (MDS). Patients with MDS have bone marrow that does not produce enough mature blood cells. This causes a lack of healthy blood cells that can function properly in the body. There are different types of MDS. REVLIMID® is for the type of MDS with a chromosome problem where part of chromosome 5 is missing. This type of MDS is known as deletion 5q MDS. Patients with this type of MDS may have low red blood cell counts that require treatment with blood transfusions.
REVLIMID® is also used with dexamethasone to treat patients with multiple myeloma who have already had another treatment. Multiple myeloma is a cancer of plasma cells. Plasma cells are found in the bone marrow. Plasma cells produce a protein called antibodies. Some antibodies can attack and kill disease causing germs. Patients with this type of cancer may have low blood cell counts and immune problems giving them a higher chance for getting infections such as pneumonia. The bones can be affected leading to bone pain and breaks (fractures).
REVLIMID® can only be:
- prescribed by healthcare providers who are registered in the RevAssist® program
- dispensed by a pharmacy that is registered in the RevAssist® program
- given to patients who are registered in the RevAssist® program and who agree to do everything required in the program
REVLIMID® has not been studied in children under 18 years of age.
Who should not take REVLIMID®?
- Do not take REVLIMID® if you are pregnant, plan to become pregnant, or become pregnant during REVLIMID® treatment. REVLIMID® may cause birth defects. See “What is the most important information I should know about REVLIMID®?”
- Do not take REVLIMID® if you are allergic to anything in it. See the end of this Medication Guide for a complete list of ingredients in REVLIMID®.
What should I tell my healthcare provider before taking REVLIMID®?
Tell your healthcare provider about all of your medical conditions, including if you:
- are pregnant or breastfeeding. REVLIMID® must not be used by women who are pregnant or breastfeeding.
Tell your healthcare provider about all the medicines you take including prescription and non-prescription medicines, vitamins and herbal supplements. It is possible that REVLIMID® and other medicines may affect each other causing serious side effects.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist.
How should I take REVLIMID®?
- Take REVLIMID® exactly as prescribed.
You must also follow all the instructions of the
RevAssist® program. Before
prescribing REVLIMID®, your healthcare
provider will:
- explain the RevAssist® program to you
- have you sign the Patient-Physician Agreement Form
You will not be prescribed REVLIMID® if you cannot agree to or follow all of the instructions of the RevAssist® program.
You will get no more than a 28-day supply of REVLIMID® at one time. This is to make sure you follow the RevAssist® program.
- Swallow REVLIMID® capsules whole with water once a day. Do not break, chew, or open your capsules.
- If you miss a dose of REVLIMID®, take it as soon as you remember that day. If you miss taking your dose for the entire day, go back to taking your regular dose the next day. Do not take 2 doses at the same time.
- If you take too much REVLIMID® or overdose, call your healthcare provider or poison control center right away.
- You will have regular blood tests during your treatment with REVLIMID®. If you are being treated for del 5q myelodysplastic syndromes (MDS) you should have your blood tested every week during your first 8 weeks of treatment, and at least monthly after that. If you are being treated for multiple myeloma, your blood counts should be checked every two weeks for the first 12 weeks and then at least monthly after that. Your healthcare provider may adjust your dose of REVLIMID® or interrupt your treatment based on the results of your blood tests and on your general condition.
- Female patients who can get pregnant will get regular
pregnancy testing.
- get a pregnancy test weekly for 4 weeks.
- Female patients who can become pregnant must agree to use 2 separate forms of effective birth control at the same time, 4 weeks before, while taking, and for 4 weeks after stopping REVLIMID®.
- Male patients, even those who have had a vasectomy, must agree to use a latex condom during sexual contact with a pregnant female or a female who can become pregnant.
What should I avoid while taking REVLIMID®?
- Do not get pregnant while taking REVLIMID® and for 4 weeks after stopping REVLIMID®. See “What is the most important information I should know about REVLIMID®?”
- Do not breastfeed while taking REVLIMID®. We do not know if REVLIMID® passes into your milk and harms your baby.
- Do not share REVLIMID® with other people. It may cause birth defects and other serious problems.
- Do not give blood while you take REVLIMID® and for 4 weeks after stopping REVLIMID®. If someone who is pregnant gets your donated blood, her baby may be exposed to REVLIMID® and may be born with birth defects.
- Male patients should not donate sperm while taking REVLIMID® and for 4 weeks after stopping REVLIMID®. If a female who is trying to become pregnant gets your sperm, her baby may be exposed to REVLIMID® and may be born with birth defects.
What are the possible side effects of REVLIMID®?
-
REVLIMID® may
cause serious side effects including:
- birth defects
- low white blood cells and platelets
- blood clots in veins and in the lungs
See “What is the most important information I should know about REVLIMID®?”
Other common side effects of REVLIMID® are:
- diarrhea
- itching
- rash
- tiredness
Tell your healthcare provider about any side effect that bothers you or that does not go away.
These are not all the side effects with REVLIMID®. Ask your healthcare provider or pharmacist for more information.
How should I store REVLIMID®?
Store REVLIMID® at room temperature, 59° to 86°F (15° to 30°C).
Keep REVLIMID® and all medicines out of the reach of children.
General information about the safe and effective use of REVLIMID®
Medicines are sometimes prescribed for conditions that are not mentioned in Medication Guides. Do not take REVLIMID® for conditions for which it was not prescribed. Do not give REVLIMID® to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide provides a summary of the most important information about REVLIMID®. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about REVLIMID® that is written for healthcare professionals. You can also call 1-888-423-5436 or visit www.REVLIMID.com.
What are the ingredients in REVLIMID®?
REVLIMID® (lenalidomide) capsules contain 5 mg, 10 mg, 15 mg or 25 mg of lenalidomide and are available as gelatin capsules for oral administration.
The inactive ingredients of REVLIMID® capsules are: lactose anhydrous, microcrystalline cellulose, croscarmellose sodium, and magnesium stearate.
The 5 mg and 25 mg capsule shell contains gelatin, titanium dioxide and black ink. The 10 mg capsule shell contains gelatin, FD&C blue #2, yellow iron oxide, titanium dioxide and black ink. The 15 mg capsule shell contains gelatin, FD&C blue#2, titanium dioxide and black ink.
Manufactured for Celgene Corporation
Summit, NJ 07901
This Medication Guide has been approved by the US Food and Drug Administration.
RevPlyMG.006 01/09
| REVLIMID
lenalidomide capsule |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| REVLIMID
lenalidomide capsule |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| REVLIMID
lenalidomide capsule |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| REVLIMID
lenalidomide capsule |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
Revised: 04/2009Celgene Corporation